INTRODUCTION
Outer dense fiber 2 (ODF2) is a double leucine zipper motif containing a 70 kDa protein. It was initially considered a major component of sperm flagella outer dense fibers (Brohmann et al., 1997; Schalles et al., 1998; Shao et al., 1997; Turner et al., 1997) and was later identified as playing a role in distal/subdistal appendages localization on the mother centriole in cultured cells (Nakagawa et al., 2001). Outer dense fibers are the main cytoskeletal structure in the mid and principal piece of sperm tails, and they protect the sperm tail and they play an important role in maintenance and elastic recoiling sperm tail (Baltz et al., 1990; Lindemann, 1996). The developmental defect of outer dense fibers causes severe sperm tail abnormalities and infertility (Haidl et al., 1991). It has been reported that tyrosine residue phosphorylation of ODF2 is important in sperm motility (Mariappa et al., 2010). The splicing variant, Cenexin1, has an additional 167 amino acid (aa) unique C-terminal extension (molecular mass of 93 kDa). Recently, it has been reported that ODF2 and its variant, Cenexin1, have different functions in male germ cells and somatic cells (Soung et al., 2009; Soung et al., 2006). ODF2 is dominantly expressed in the testis from pachytene spermatocytes (Horowitz et al., 2005; Turner et al., 1997), whereas Cenexin1 is abundantly expressed in the somatic tissues. Furthermore, Cenexin1 has an important function in ciliogenesis and cell cycle progression at the centrosome while ODF2 does not (Soung et al., 2009). In the early cell cycle stages, Cenexin1 recruits ninein to the centrosome and facilitates ciliogenesis in hTERT-RPE cells. In late G2 and M phase, Cenexin1 recruits Plk1 to the centrosome to make proper bipolar spindle. Phosphorylation of Cenexin1 S796 by Cdc2 is important for Plk1 binding, and it results in the proper targeting of Cenexin1 protein to its mother centriole, which is critical for mitotic progression (Soung et al., 2009). In the genetic model RO072 ES cell-derived ODF2–/– knockout mice incur embryonic lethality before the blastocyst stage, but there are no abnormalities in ODF2+/– mice (Salmon et al., 2006). Whereas, XL169 ES cell derived heterozygote mice present severe male infertility and abnormal sperm tail structures (Tarnasky et al., 2010). Until now, there has been no in vivo study examining the role of Cenexin1 S796 phosphorylation and the functional difference between ODF2 and Cenexin1. In this study, to elucidate in vivo meaning of Plk1 binding to Cenexin1, we generated Cenexin1 S796A mutants then we found that the ectopic expression of Cenexin1 S796A, which is an incompetent mutant for Plk1 binding, in RO072 ES cell derived ODF2 heterozygote background causes severe defects in sperm tail development, while the ectopic expression of ODF2/Cenexin1 WT does not.
MATERIALS AND METHODS
The KpnI-SalI fragments of each Cenexin1 WT, Cenexin1 S796A, or ODF2 were amplified from pShuttle-CMV-EGFP-Cenexin1 WT (lab stock number of M674), pShuttle-CMV-EGFP-Cenexin1 S796A (lab stock number of M675) or pUC19-ODF2 (lab stock number of M621) by PCR. All PCR products were confirmed by sequencing analysis. Each PCR product was double digested with KpnI and SalI restriction enzymes, and then subcloned into the KpnI–XhoI site of pShuttle-Flag vec (Flag sequence was inserted into the AgeI-BglII site of pShuttle CMV vector, Stratagene, Cedar Creek, TX). Each flag-tagged cDNA was excised from a pShuttle-Flag-Cenexin1 WT/Cenexin1 S796A/ODF2 using AgeI and EcoRV double digestion. Both excised cDNAs and BglII-XhoI digested PCCALL2-anton ΔlacZ vectors (a gift from Michael Bustin, NIH/NCI, Bethesda, MD) were end filled by Klenow (New England Biolabs, Ipswich, MA) reaction including dNTPs. Blunt end cDNAs were subcloned into end-filled PCCALL2-anton ΔlacZ vector.
Cenexin1 WT/Cenexin1 S796A/ODF2c DNA was cloned into the PCCALL2-anton ΔlacZ vector, as described above. The sequence and orientation were verified using an ABI Prism Big Dye Terminator Kit (Applied Biosystems, Foster City, CA). The Cenexin1 WT/Cenexin1 S796A/ ODF2 transgene was excised from the vector by BamHI-ScaI digestion and purified before microinjection into fertilized C57BL/6 mouse eggs. Transgene-positive mice were screened by Southern blot analysis and mated to ODF2+/– mice.
Southern blot analysis was performed by a standard protocol as described previously (Chiang et al., 2000). Mouse tail genomic DNA from wild-type or transgenic mice was digested with BamHI, electrophoresed on a 0.7% agarose gel, and transferred onto nylon filters. The filters were hybridized with 32P-labeled probe in Hyb buffer (Stratagene) at 65°C for 4 to 8 h. The probe was labeled with [α-32P]dCTP with a room temperature random primer kit (Stratagene). Filters were exposed after 3 washes with 0.2× SSC (1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate) and 0.2% sodium dodecyl sulfate for 10 min at 60°C.
To detect 676-bp wild-type and 147-bp mutant ODF2 allele specific PCR products, we followed previously described methods (Salmon et al., 2006). Briefly, genomic DNA was isolated from mouse tail and each wild type and mutant allele was amplified using a common forward primer 5 -CCGAGAGACTAATGGAGCAAC-′ 3′; wild type reverse 5′-CTGGTCCACTTCGCTCTCTC-3′; and mutant reverse primer 5′-CCACAACGGGTTCTTCTGTT-3′. For the PCR reaction, we used 95°C for 3 min denaturation and 35 cycles of 95°C for 20 s, 58°C for 20 s, and 72°C for 30 s. PCR products were electrophoresed in 1% agarose gel stained with ethidium bromide.
To detect expression of transgenes from mouse tail, the mouse tail was homogenized and lysed in RIPA buffer (50 mM Tris-Cl (pH 7.4), 150 mM NaCl, 1% NP40, 0.5% Na-deoxycholate, 0.1% SDS, 1X protease inhibitor cocktail, and 20 mM p-nitrophenyl phosphate) and briefly centrifuged. A 2X SDS sample buffer was added to the supernatant and boiled. Samples were subjected to 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) followed by western blot analysis using anti-Cenexin1/ODF2 antibody. For immunoprecipitation, various mouse tissues were homogenized and lysed in RIPA buffer. Tissue lysates were centrifuged at 4°C, 14,000 rpm for 20 min. The supernatants were incubated with anti-Flag antibody conjugated beads (Sigma, St. Louis, MO) for 4 h at 4°C. Beads were washed with 1X RIPA buffer for 4 times and then boiled after adding 2X SDS sample loading buffer. Protein samples were separated by 10% SDS-PAGE and transferred to PVDF membranes. Membranes were incubated with anti-Cenexin1/ODF2 antibody for 2 h at room temperature followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (Amersham Biosciences, Piscataway, NJ) incubation. Immunoreactive signals were detected by an enhanced chemiluminescence (ECL) detection system (Pierce, Rockford, IL).
Mouse testes were fixed in formalin solution (Sigma) and embedded in paraffin. For hematoxylin & eosin staining, sections had the paraffin removed in a SAFECLEAR II tissue-clearing agent (Fisher Scientific, Kalamazoo, MI), rehydrated in a series of ethanol solutions, and stained with hematoxylin and eosin solution (Sigma). For immnohistochemistry, rehydrated testes sections were repeatedly subjected to 3 cycles of boiling and cooling in 10 mM citric acid solution (pH 6.0) for antigen retrieval, and then probed with primary antibodies against acetylated- α-tubulin (Sigma) and Cenexin1/ODF2 (in this study) for overnight at 4°C and followed by Texas-Red/Alexa flour 488-conjugated secondary antibody (Invitrogen, Carlsbad, CA) incubation for 1 h at room temperature. DNA was stained with 0.1 μg/ml of 4′,6′-diamidino-2-phenylindole (DAPI) (Sigma) for 10 min in 1X PBS diluted DAPI solution. Fluorescence signals were observed under Zeiss LSM510 confocal microscope.
RESULTS
To investigate in vivo meaning of Plk1 binding to Cenexin1, we generated Cenexin1 S796A mutants, which are incompetent mutants in Plk1 binding, expressing transgenic mice by using an ODF2+/– knock out background. To examine the functional differences between Cenexin1 and ODF2, we also generated Cenexin1 WT/ODF2 expressing transgenic mice by using the same strategy as the Cenexin1 S796A transgenic mice. To generate transgenic mice Flag-hODF2, Flag-hCenexin1 S796A mutant, or Flag-hCenexin1 wild type (WT) cDNA was subcloned into the PCCALL2-anton ΔlacZ vector as described in the Materials and Methods and Fig. 1a.
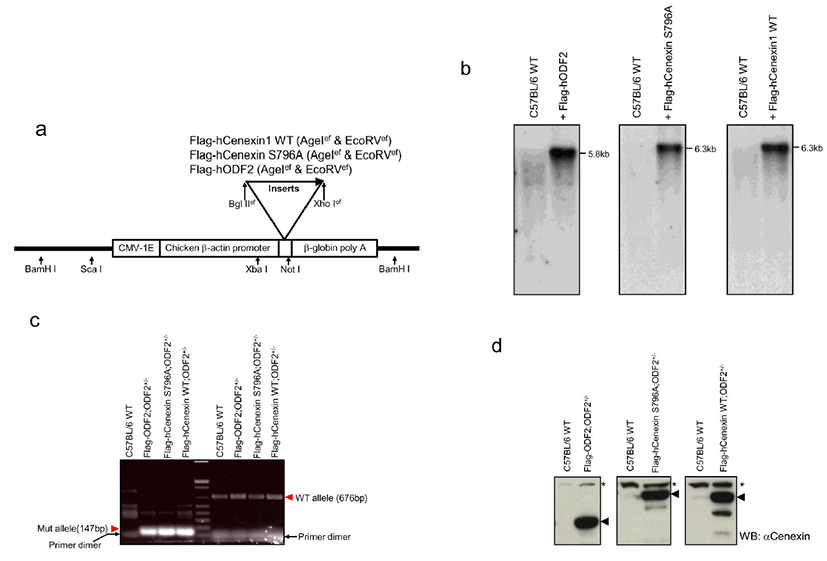
Integration of the transgene into genomic DNA was identified by Southern blot analysis (Fig. 1b). The BamHI digestion of genomic DNA produced a 5.8 kb or 6.3 kb transgenic vector fragment, including 1.9 kb of ODF2 (GenBank accession number AF012549) or 2.4 kb of Cenexin1 WT/S796A (GenBank accession number DQ444714) inserts, respectively (Fig. 1b). In 3 different gene-expressing mice, we established 2 transgene-transmitted lines. We then mated each transgenic mouse with an ODF2+/– mouse to produce a transgene possessing ODF2+/– mouse. We genotyped each mouse (Fig. 1c) and checked for the expression of the transgene (Fig. 1d). For allele genotyping, we isolated genomic DNA from mouse tail snip and carried out PCR analysis by using a common forward primer and each allele-specific reverse primer (Salmon et al., 2006). We confirmed the 676 bp wild-type and 147 bp mutant allele PCR products from each transgenic line (Fig. 1c) and confirmed all 3 transgene expressions from tail extracted lysate through western blot analysis by using anti-Cenexin1/ODF2 antibody in each transgenic line (Fig. 1d). Although anti-Cenexin1/ODF2 antibody recognized both ODF2 and its splicing variant Cenexin1 at an endogenous level, only the expression of the transgene was observed from the tail extract in each transgenic mouse compared with normal C57BL/6 wild-type (Fig. 1d).
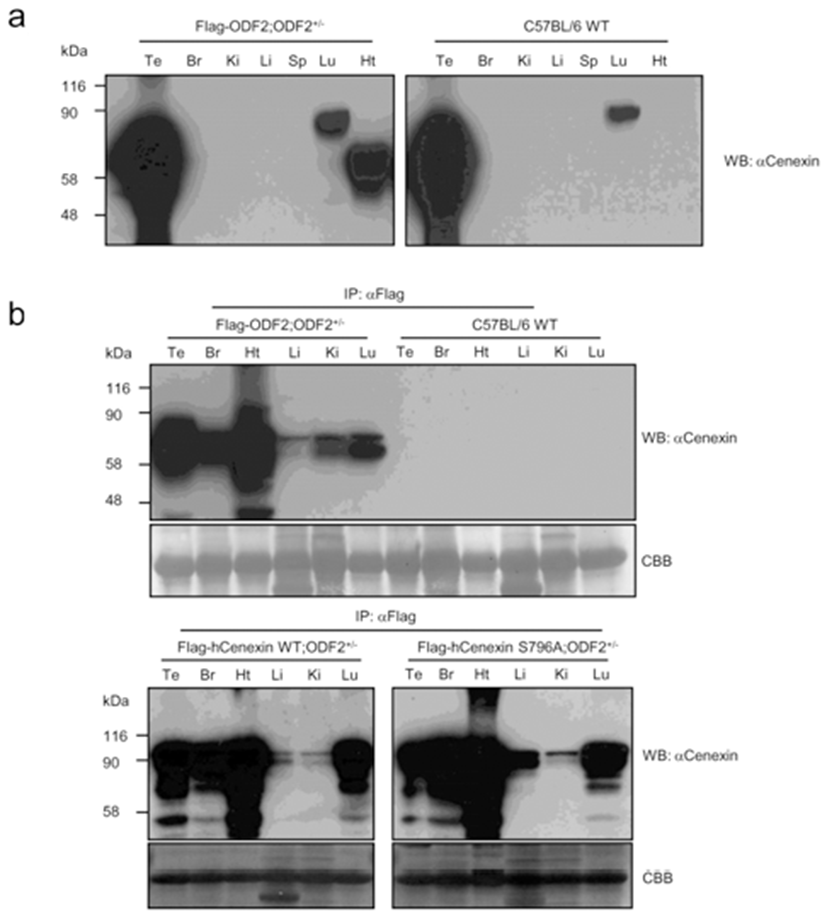
We presume that, unlike cultured cell lines, mouse tails contain a low level of endogenous Cenexin1 protein. We then checked the distribution of transgene expression among the tissues. First, we detected the expression of transgenes from various tissue lysates through conventional western blot analysis. In comparison with the C57BL/6 WT mouse (Fig. 2a), we observed specific expression of the transgene in the heart and a large amount of endogenous expression of ODF2 and Cenexin1 in the testis and lung, respectively. We could not detect any immunized signal from other tissues, such as brain, kidney, liver, and spleen (Fig. 2a). Since the β-actin promoter has ubiquitous expression characteristics, transgenes should be expressed in a variety of tissues. The reason we could not detect a ubiquitous expression is likely that transgenes may have a low expression level compared with endogenous ODF2 protein in the testis (Fig. 2a). To overcome this, we conducted immunoprecipitation assays followed by western blot analysis to detect the expression of the transgene in various tissues. We performed immunoprecipitation using an anti-Flag antibody to gather more Flag-tagged transgenic proteins from each tissue extracted lysates, followed by a western blot analysis using the anti-Cenexin1/ODF2 antibody (Fig. 2b). We observed a ubiquitous expression pattern of each transgene from this analysis (Fig. 2b), with a somewhat different expression level among tissues. All three transgenes were abundantly expressed in the heart and to a lesser degree in the liver and kidney (Fig. 2b). This contrasted with normal C57BL/6 wild type mouse tissues where there were no immunized signals in anti-Flag antibody immunoprecipitation (Fig. 2b).
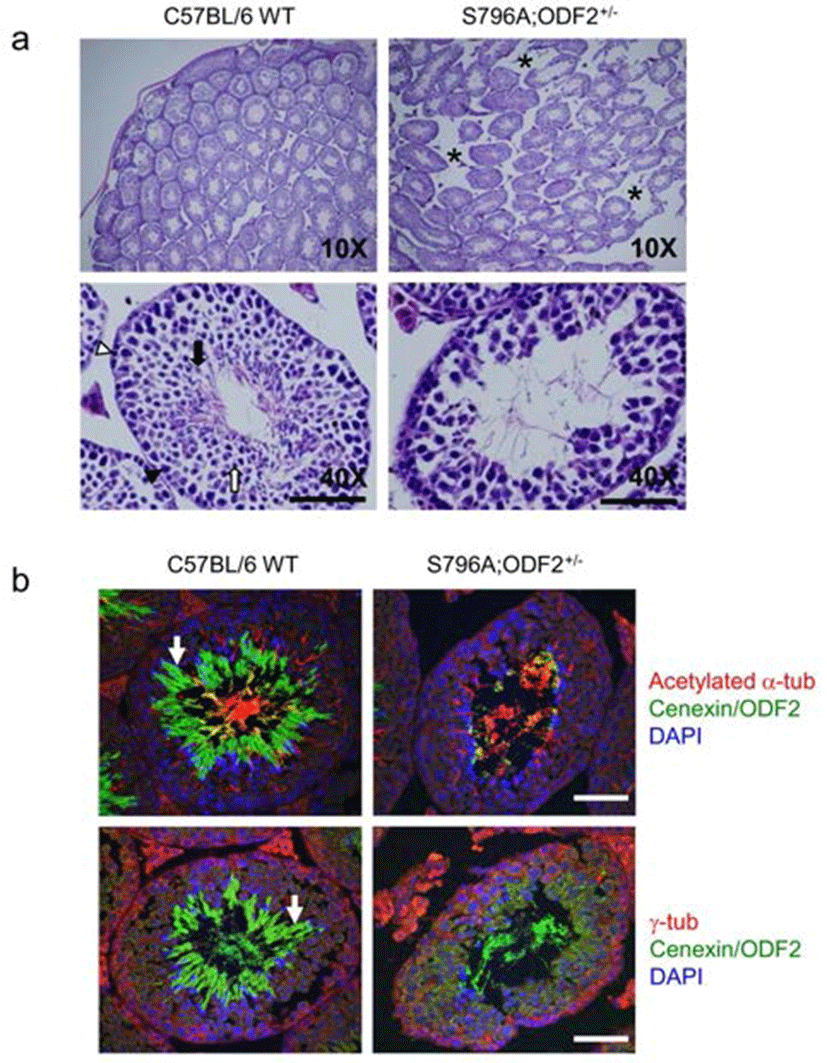
In both categories, one being transgene-expressing pups and the other is ODF2 heterozygote allele containing transgene-expressing pups, we observed that the exogenous expression of Cenexin1 S796A mutant caused a severe defect in fertility. We sacrificed most male defective-phenotypes to analyze its testicular structure through histological analysis. From hematoxylin-eosin (H & E) staining of testes, the structure of seminiferous tubules in the S796A;ODF2+/– mouse was distinguishable from that of the WT mouse. Tubules of WT mouse were compactly well arranged, while that of S796A;ODF2+/– mouse had a wider interstitial region (Fig. 3a, asterisks). Magnification of the specimen revealed that spermatogonia in the seminiferous tubules of the S796A;ODF2+/– mouse differentiated until round spermatids, but failed to accomplish morphological transition into elongated spermatids. Moreover, tubules of S796A;ODF2+/– mouse showed severe diminishment of total germ cell number (Fig. 3a) compared with WT mouse. Additionally, we found that the majority of Cenexin1/ODF2 protein localized in the tail of elongated spermatids (Fig. 3b, arrow, green) of C57BL/6WT mouse, but not in the S796A; ODF2+/– mouse. In S796A;ODF2+/– mouse, there is no tail-like structure in the testis (Fig. 3b, arrow).
DISCUSSION
In this study, we examined the in vivo functional difference between Cenexin1 and ODF2, and did not see any significant differences between Cenexin1 WT and ODF2 over-expressing transgenic mice with an ODF2+/– background. The two mouse lines were viable and fertile. However, we found an unexpected phenotype in Cenexin1 S796A mutant expressing transgenic line: Cenexin1 S796A expressing mice showed less than half the number of progeny compared with Cenexin1 WT/ODF2 transgenic mice. According to the pup number, there were no significant differences in the fertility between Cenexin1 WT/ODF2 transgenic mice and normal C57BL/6 wild-type mice. Only Cenexin1 S796A mutant mice showed a defect in fertility. After examining the most severe defective-phenotype possessing male mice, we found that there was a severe defect in spermiogenesis. Specifically, there was a defect in the differentiation from round spermatids to elongated spermatids according to the histological analysis. Furthermore, the total germ cell number and structure of the seminiferous tubule are quite different from those of normal C57BL/6 wild type mouse. Previously, it was reported that Cenexin1/ODF2 localized at sperm tail and it may be important for the proper differentiation of spermatozoa (Oko, 1988; Schalles et al., 1998). Thus, it is possible that Plk1 binding defective-mutant Cenexin1 protein, Cenexin1 S796A, does not localize at the proper site, which is important for the differentiation of the sperm tail.
One of the initial goals was to identify the in vivo functional difference between Cenexin1 and ODF2. We attempted to express Cenexin1 or ODF2 transgenes in a ODF2–/– knockout background, but this was unsuccessful. This suggests that the introduction of transgene, even ODF2, did not make enough functional proteins to rescue the embryonic lethal phenotype of the homozygote ODF2 knockout mouse. Additionally, in natural conditions, ODF2 is dominantly expressed in the testis (Horowitz et al., 2005; Turner et al., 1997; Hoyer-Fender et al., 1998; Soung et al., 2009). Thus, to properly recover the endogenous ODF2 function in the developmental stage, it is better to use a testisspecific expression system than an ubiquitous expression system.
We observed a severe defect in mouse sperm tail development in Cenexin1 S796A mutant expressing mouse in the ODF2+/– background. From this observation, we suggest it is possible to maintain proper function of ODF2 in the testis, the introduction of Cenexin1 needs Plk1 binding or proper structural changes through the phosphorylation of S796 site. These data also suggest the ectopic expression of Cenexin1 S796A, phosphorylation-/Plk1 binding-deficient mutant, may interrupt the proper localization or function of endogenous ODF2 protein in the differentiation process of sperm tails. Because we could not get ODF2–/– background mouse, we cannot exclude the possibility that this phenotype arises from a combination effect of both exogenous Cenexin1 S796A and endogenous ODF2 proteins in ODF2+/– background mouse.
Recently Tarnasky et al. (2010) reported that the XL169 ODF2+/– ES cell line oriented chimeric male mice were all infertile and that their epididymal spermatozoa presented tail defects. Even though the authors did not introduce transgenes into the ODF2+/– mice, those phenotypes are consistent with our results. Collectively, these results suggest it may be possible that Cenexin1/ODF2 has an important function in spermatozoa tail development and ectopic expression of poorly phosphorylated Cenexin1 or Plk1 unbound Cenexin1 in the testis interrupt spermatogenesis.