
INTRODUCTION
Autophagy is a multistep catabolic process that occurs during nutrient deprivation, metabolic stresses, tumor growth, normal mammalian development and differentiation (Degenhardt et al., 2006, Mizushima & Levine, 2010). It begins with the formation of double-membrane vesicles, autophagosomes, which engulf cytoplasmic constituents. The autophagosomes fuse with lysosomes, where the damaged or long-lived proteins and organelles are degraded and recycled as a protective mechanism and adaptive response to stressful conditions (Degenhardt et al., 2006). Excessive and sustained activation of autophagy, however, seems to provoke cell demise (autophagic cell death) by depleting cell organelles and critical proteins. Consistent with its function to either induce cell death or promote cell survival, several lines of evidence suggest the paradoxical role of autophagy in cancer cells induced by a variety of anti-cancer drugs, with response enhancing or counteracting their anticancer effect (Degenhardt et al., 2006).
Chloroquine (CQ) is one of lysosomotropic agents as a weak basic amine. It can freely diffuse across lysosomal membranes in uncharged form but can also become protonated and trapped within acidic vesicles such as lysosomes. The protonation of CQ results in inhibition of hydrolytic lysosomal enzymes by perturbing the acidic milieu (pH<5) of lysosome, which has made CQ an useful drug in the treatment of malaria (Kroemer & Jaattela, 2005). CQ can suppress autophagy by accumulating in the lysosomal lumen and inhibiting the autophagolysosome formation (Boya & Kroemer, 2008). Since several current studies have revealed that tumor resistance to anticancer therapies including radiation therapy, chemotherapy and molecular targeted therapies is attributed to upregulation of autophagy as a protective mechanism (Hu et al., 2012; Zou et al., 2012), CQ and its derivative hyroxychloroquine have been used as an autophagy inhibitor available for clinical trials of cancer patients. In addition, inhibition of autophagy by CQ synergistically augments cytotoxicity in combination with several anticancer drugs in preclinical models (Takeuchi et al., 2005; Syelo et al., 2006; Carew et al., 2007; Xu et al., 2011; Firat et al., 2012). Recently, however, evidence has been accumulated suggesting that the ability of CQ to inhibit autophagy by blocking autophagolysosome formation may not be the only mechanism by which it exerts antitumor effect. Lysosomotropic CQ sensitizes breast cancer cells to chemotherapy independent of autophagy since autophagy blockage by knocking out autophagy gene such as Atg12 and Beclin 1, or bafilomycin A1 treatment did not show any sensitization effect to chemotherapy (Maycotte et al., 2012). Another report also showed that PI3K/mTOR inhibitor PI103 enhances the lysosomal compartment by increasing its size and function, while CQ destabilizes lysosomal membranes (Enzenmuller et al., 2013). When the accumulation of protonated CQ within lysosomes reaches above a certain threshold, it attains the detergentlike activity and leads to lysosomal membrane permeabilization (LMP) (Boya & Kroemer, 2008). LMP releases the cathepsins and other hydrolytic enzymes from lysosomal lumen into the cytosol, which can result in apoptosis with mitochondrial outer membrane permeabilization and caspases activation, or in necrosis depending on the extent of LMP and the cell context (Kroemer & Jaattela, 2005; Boya & Kroemer, 2008).
Thus the present study was undertaken to demonstrate the properties and underlying mechanism for the dual activities of CQ, autophagy inhibitor or LMP inducer, by using human colorectal cancer cell line HCT15.
MATERIALS & METHODS
The human colorectal cancer cell line HCT15 was obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in a monolayer culture in DMEM (Sigma) supplemented with 10% fetal bovine serum (Sigma) and 1% streptomycin/penicillin, and were maintained at 37°C in a humidified atmosphere consisting of 5 % CO2 and 95% air. Cells were regularly tested for mycoplasma contamination by treating 5 ug/mL of Plasmocin (InvivoGen). NVP-BEZ235 was purchased from LC laboratories (Woburn, MA). CQ, N-acetylcysteine (NAC), trolox, acridine orange, 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) and Hoechst33342 were all purchased from Sigma and Image-iT LIVE Green Reactive Oxygen Species Detection Kit was from Molecular Probes.
To determine the effects of CQ and/or NVP-BEZ235 on HCT15 cell viability after the treatments, we used MTT assay as described previously (Song et al., 2011). Cells were harvested and seeded at 4 × 104 cells per well (0.5 mL) in 24-well plates and incubated overnight at 37°C. The cells were then incubated for 48 hours with CQ and/or NVP-BEZ235, or for 12, 24, 48, and 72 hours with CQ to determine the dose- and time-dependent cell survival rate. Then the cells were incubated with MTT reagent for 3 hours at 37°C, followed by solubilization of the formazan crystal with propanol for 30 minutes. Absorbance was measured at 570 nm with a microplate analyzer. All the experiments were performed in triplicate.
The characteristics of autophagy is the formation of acidic vesicular organelle (AVO) which consist predominantly of autophagosome and autophagolysosome. To visualize the development of AVOs after CQ treatment, we performed vital staining with acridine orange. Briefly cells were washed twice with D-PBS, stained with acridine orange (1 ug/mL) in HBSS containing 5% FBS for 15 minutes and then observed with a fluorescence microscope.
RESULTS
To test the effect of CQ on the survival of colorectal cancer cells, we treated HCT15 cell line harboring PI3K and K-Ras mutation with CQ at concentrations of 5 uM to 80 uM for 12, 24, 48, or 72 hours and then antiproliferative activity of CQ was measured by using MTT assay. As shown in Fig. 1, CQ inhibited cell viability in dose- and time-dependent manners in the range of CQ concentration between 20 to 80 uM, whereas it did not show antiproliferative activity at 5 and 10 uM.
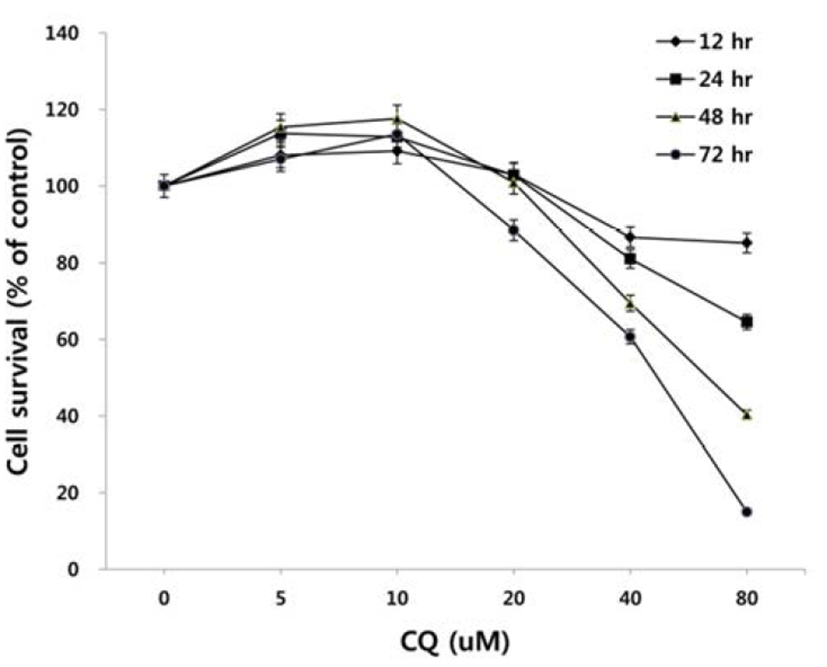
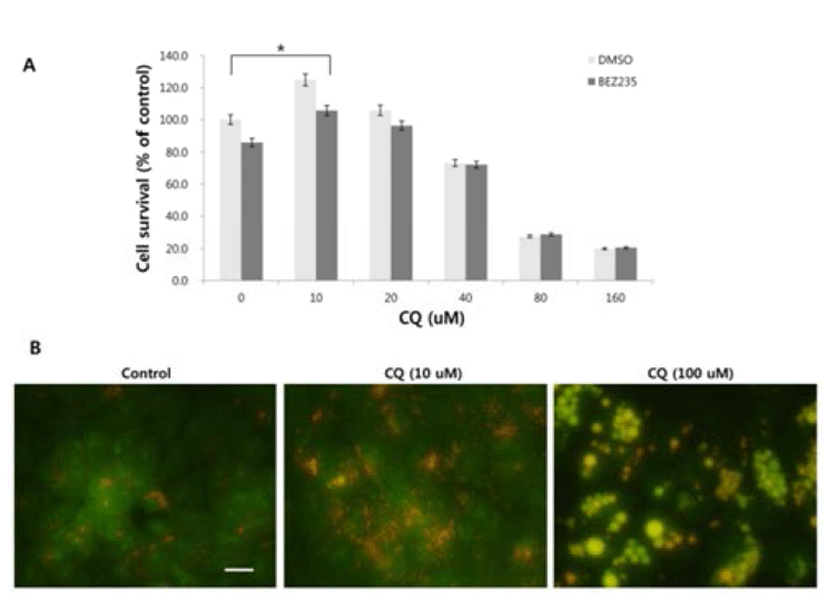
There has been some conflicting results and interpretation about the effects of CQ treatment on cancer cells, which seem to be due to the varied concentrations of CQ treated (Xu et al., 2011; Seitz et al., 2013). CQ is not only an autophagy inhibitor by blocking the fusion of autophagosome and lysosome in the late stage of autophagy, but it can lead to cell death since the protonated form of CQ reaching above a certain threshold acquires detergent-like properties, which results in lethal lysosomal destabilization known as LMP (Boya & Kroemer, 2008). Therefore, we hypothesized that CQ may act as an autophagy inhibitor at a low concentration, but as an LMP inducer at a high concentration above the threshold attaining detergent-like activity. To prove this, we employed HCT15 cell line, since our unpublished study showed that autophagy inhibition by CQ rescued the HCT15 cell viability in cotreatment regimen with anticancer drug NVP-BEZ235 (BEZ235) of a dual inhibitor of PI3K and mTOR (Maira et al., 2008). As revealed in Fig. 2A, CQ at low concentrations (10–20 uM) seemed to exert its effect as an autophagy inhibitor by rescuing the cell viability by both CQ treatment and cotreatment with CQ and NVP-BEZ235, while it induced the lethal effect at high concentrations (40–160 uM). Next, we tried to monitor the development of AVOs as a function of CQ concentration by staining cells with acridine orange. Acridine orange (AO) is a lysosomotropic metachromatic fluorochrome, which emits red fluorescence at high concentrations when in lysosomes and green fluorescence at low concentrations in cytoplasm and nucleus. Therefore, reduced red fluorescence in lysosomes of AO-loaded cells can reflect LMP (Antunes et al., 2001). The untreated HCT15 tumor cells showed little cytoplasmic staining. Notably, treatment of the tumor cells with a low concentration (10 uM) of CQ exhibited the development of abundant red fluorescent AVOs, revealing autophagy inhibition effect. Meanwhile, reduced red or green fluorescence together with enlargement of AVOs was observed after applying a high concentration of CQ treatment, raising the possibility of LMP induction (Fig. 2B).
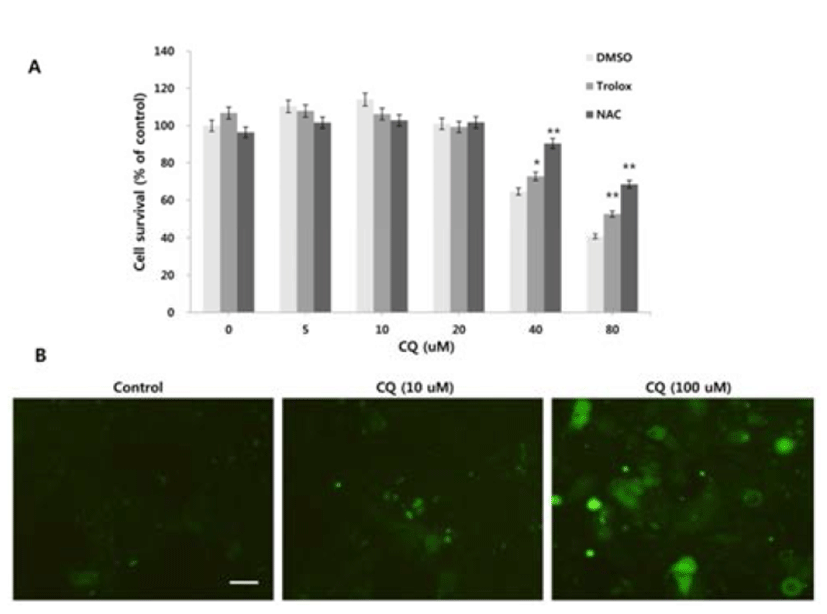
Since reactive oxygen species (ROS) can regulate the induction of autophagy and LMP (Terman & Kurz, 2006; Scherz-Schouval et al., 2007), we investigated whether ROS is generated in response to the high concentration of CQ treatment and whether ROS production contributes to the cell viability. We used the general ROS scavenger N-acetylcysteine (NAC), a precursor of the antioxidant glutathione that has antioxidant effects via its thiol group (Zafarullah et al., 2006), and trolox, a derivative of the antioxidant vitamine E (Seifried et al., 2003). At high concentrations of CQ, both NAC and trolox resulted in the rescue of cell viability (Fig. 3A). We next analyzed the generation of ROS upon treatment of CQ using the Image-iT Live green ROS detection kit. Increased cellular ROS were detected following high concentration of CQ treatment, but the low concentration of CQ treatment showed little ROS positive signal (Fig. 3B).
DISCUSSION
The present study, using human colon cancer cell line HCT15 harboring PIK3CA and K-Ras mutations, was undertaken to evaluate the nature of anticancer effect of CQ. Our results show that CQ has biphasic activity depending on its concentrations treated; CQ at low concentrations seems to act as an autophagy inhibitor while acting as an LMP inducer at high concentrations. In addition, treatment of CQ leads to increased generation of ROS. Also, addition of antioxidants such as NAC and trolox rescues the tumor cell viability in response to high doses of CQ.
We first screened the antiproliferative effect of CQ by MTT assay. Our results revealed that exposure of HCT15 cancer cells to CQ inhibits the cell viability in a dose- and time-dependent manner. Interestingly, a low concentration of CQ treatment increases cell survival. Cancer cells activate autophagy in response to high metabolic rate associated with rapid cell proliferation (Yang et al., 2011). Human cancer cell lines with activating mutations in Ras commonly have high levels of basal autophagy even in the presence of abundant nutrients (Guo et al., 2011). Moreover, the expression of oncogenically mutated Ras gene induces cell death with morphological and biochemical characteristics typical of autophagic cell death in human glioma and gastric cancer cell line (Chi et al., 1999). The persistent and excessive autophagy can lead to autophagic cell death (type II physiological cell death) depending on the cellular context (Li et al., 2013). Thus, considering that the colorectal cancer cell line of HCT15 includes K-Ras mutation in addition to phosphoinositide-3-kinase catalytic alpha polypeptide (PIK3CA) mutation, the autophagic cell death of tumor cells may manifest in the normal replete conditions, which is suppressed by low concentrations of CQ. This notion is further confirmed by the combination treatment of CQ with antitumor agent BEZ235. Autophagy is negatively regulated by mTOR and inhibition of mTOR with BEZ235 stimulates the autophagy in cancer cells, in which autophagy acts in either prodeath or prosurvival manners (Xu et al., 2011; Li et al., 2013). Our unpublished data showed that cotreatment of BEZ235 with low doses of CQ (5–20 uM) rescues the viability of HCT15 cancer cells, which enables us to use as an experimental model to delineate the dosedependent dual activities of CQ, i.e., autophagy inhibitor or LMP inducer in a dose-dependent manner. Our present data indicate that CQ at low concentrations counteracts the antitumor effect of BEZ235, while high concentrations of CQ results in LMP induction. This is consistent with a recent study using a relatively high concentration of CQ (50 uM) that cotreatment of BEZ235 with CQ triggers LMP and apoptosis via mitochondrial-lysosomal cross-talk (Seitz et al., 2013). They demonstrate that inhibition of BEZ235-stimulated autophagy by silencing or knockdown of autophagy-related genes such as Atg7 or Atg5 do not alter the induction of apoptosis by the combination treatment with BEZ235 plus CQ. Together, our present results and others suggest that the outcome of CQ cotreatment with other anticancer drugs might not be due to the CQ effect on autophagy induced by the chemotherapeutic agents, as the effect may be resulted from mechanisms other than its autophagy inhibition, such as LMP induction.
Recently, it has been suggested that ROS results in induction of autophagy and LMP (Terman & Kurz, 2006; Scherz-Schouval et al., 2007). The present study is, to our knowledge, the first report to show production of cellular ROS in response to CQ in colorectal cancer cells although underlying mechanism of CQ-mediated ROS producion is unclear. A number of cancer cell lines, including colon cancer cells, have elevated steady state levels of mitochondrial superoxide relative to normal epithelial cells (Aykin-Burns, et al., 2009). Superoxide does not readily traverse the mitochondrial membrane and the decreased manganese superoxide dismutase (SOD) activity and expression have been reported in colorectal cancer cells (Bernard et al., 1997), leading to the accumulation of superoxide in the mitochondria. It is, therefore, plausible that the increased permeability of mitochondrial membrane by cathepsins liberated from lysosome after LMP induction in response to CQ treatment may be responsible for the translocation of mitochondrial superoxide to cytoplasm, where it is converted to H2O2 and other ROS by cytoplasmic copper/ zinc SOD. However, this speculation awaits further experimental verification.
Taken together, the present study suggests that low doses of CQ exert its effect as an autophagic inhibitor and CQ at high doses acts as an LMP inducer leading to cell death. In addition, there may be three possible modes for the anticancer effect by high doses of CQ in HCT15 colorectal cancer cells: 1) by LMP induction by detergentlike activity of protonated CQ that is accumulated above a certain threshold in lysosomes, 2) by autophagic cell death due to excessive and sustained autophagy, which is attributed to the high level of basal autophagy by Ras mutation in HCT15 cell line and additional induction of autophagy in response to ROS produced by CQ treatment, 3) by LMP induction in response to ROS produced by CQ treatment. However, our finding on the biphasic activities of CQ needs further investigation for the ongoing clinical trials where CQ is used simply as an autophagy inhibitor.