
INTRODUCTION
Arsenic is a naturally occurring element that exists ubiquitously in the environment in both organic and inorganic forms combined with other elements such as oxygen, chlorine, and sulfur, as well as carbon and hydrogen (Cullen and Reimer, 1989; WHO, 2000; IARC, 2004). It is classified chemically as a metalloid, having properties intermediate between a metal and a nonmetal; however, it is frequently referred to as a metal (IARC, 1980). Elemental arsenic is the 20th most common in the earth’s crust, and is emitted to the environment as a result of volcanic and industrial activities. Mining, smelting of non-ferrous metals and burning of fossil fuels are the major anthropogenic sources of arsenic contamination of air, water, and soil. Arsenic and arsenic compounds are used in pharmaceuticals, wood preservatives, agricultural chemicals (e.g. pesticides, insecticides, and herbicides), and applications in the mining, metallurgical, glass-making, and semiconductor industries (Mandal, 2002). Especially, the historical usage of arsenic-containing pesticides and wood preservatives led to contamination of the agricultural land and groundwater (WHO, 2001). Human exposure to arsenic has become a major global health problem because the worldwide contaminations and adverse health effects. In most populations, the main source of arsenic exposure is the drinking water (NRC, 1999, 2001; Smith et al., 2002; Watanabe et al., 2003). In drinking water, the current maximum contamination levels of arsenic were lowered from 50 μg/L to 10 μg/L by USEPA and WHO (IARC, 2004). Arsenic in drinking water is mainly inorganic arsenic and more harmful than in food such as grains and vegetables (Akter et al., 2005). Moreover, inorganic arsenic is more toxic than organic arsenic and can exist in two major oxidation states: a trivalent form, arsenite (As3+), and a pentavalent form, arsenate (As5+). Biologically, arsenite is 2~10-fold more active and toxic than arsenate (Kosentt, 1994). Of the inorganic arsenic compounds, arsenic trioxide (As2O3), sodium arsenite (NaAsO2) and arsenic trichloride (AsCl3) are the most common trivalent compounds, and arsenic pentoxide (As2O5), arsenic acid (AsH3O4) and arsenates [e.g. sodium arsenate (Na2HAsO4), lead arsenate (PbHAsO4) and calcium arsenate (As2Ca3O8)] are most common pentavalent compounds.
The toxic effects of arsenic that are of most concern to humans are those that occur from chronic low-level exposure. Epidemiological studies have indicated that chronic exposure to arsenic is associated with increased risks of various cancers, including those of the skin, lung, bladder, kidney, and liver, as well as numerous other non-cancer illnesses including vascular and cardiovascular disease, diabetes, reproductive and developmental problems, and neurologic and cognitive problems (Abernathy et al., 2003’ NRC 1999, 2001; Wasserman et al., 2004; Watanabe et al., 2003). Conversely, arsenic has been considered as an effective chemotherapeutic agent in the treatment of certain human cancers (Douer & Tallman, 2005). Among arsenic compounds, arsenic trioxide (As2O3) has successfully been employed in the treatment of acute promyelocytic leukemia (APL) (Soignet et al., 1998). It has also been shown that As2O3 efficiently induces apoptosis in malignant APL cells in vitro (Hu et al., 1999). These observations has been confirmed that As2O3 can induce apoptosis in other leukemia cells and solid tumor cells, including gastric cancer cells, neuroblastoma cells, and prostate and ovarian carcinomas (Zhang el al., 1999; Uslu et al., 2000). However, arsenic is commonly regarded as a potent carcinogen that can induce the formation of various types of solid tumors (Smith et al., 1992; Liu & Waalkes, 2008). Our previously study demonstrated that As2O3 effectively provokes cytotoxicity and apoptotic cell death in mouse testicular Sertoli cells in vitro (Kim et al., 2011). These investigations indicates that the male reproductive toxicity of arsenic may be directly associated with damage of testicular cells.
In addition, various experimental models have been developed to understand how arsenic exposure causes these diverse disease outcomes. However, the reproductive and developmental toxicity of arsenic is poorly understood, and specific relationships between experimental and human exposures are not established. Several epidemiologic studies have reported that arsenic exposure in utero increased spontaneous abortion and stillbirth and decreased birth weight (Ihrig et al., 1998; Ahmad et al., 2001; Milton et al., 2005). However, these studies lack detailed information about exact effects of maternal arsenic exposure. The reproductive and developmental toxicity of arsenic have primarily been documented through murine studies, suggesting arsenic as a risk to the developing fetus. In animals, some experimental studies have reported that arsenic intoxication is associated with spermatotoxicity (Waalkes et al., 2003; Pant et al., 2004), inhibition of testicular steroidogenesis and reduction of the weight of the testes and accessory sex organs (Sarker et al., 2003). Arsenic exposure also has been associated with inhibition of ovarian steroidogenesis and gonadotrophins secretion (Chattopadhyay et al., 1999), elevation of adrenocortical steroidogenesis and plasma corticosterone level (Ghosh et al., 1999), as well as with severe metabolic disorders such as diabetes in humans (Tseng et al., 2002). However, the actual molecular events resulting in reproductive and developmental toxicity from exposure of arsenic remain unclear.
The purpose of this paper is to review and summarize the major scientific developments on the topic of reproductive and developmental toxicity associated with arsenic exposure, integrating evidence from epidemiological and experimental studies.
ARSENIC EXPOSURE TO HUMAN
Arsenic is a major global health concern throughout the world widely based on its carcinogenic potential after occupational or environmental exposure (IARC, 1987; NRC, 1999). Arsenic is commonly present in air, water, and soil and could find their routes into the human body through inhalation, ingestion and skin absorption. The primary route of arsenic exposure for the most population is via the ingestion of contaminated food or water. Inhalation or skin absorption of arsenic from contaminated sources is a minor exposure route for the general population (ATSDR 2007). Most ingested and inhaled arsenic is well absorbed through the gastrointestinal tract and lung into the blood stream. Arsenic enters into the human body through these routes, and is distributed in a large number of organs including the lung, liver, spleen, kidney, intestine, skin, and vascular and lymphatic systems, as well as reproductive and nervous systems (Hunter et al., 1942; ATSDR, 2007). After absorption through lung and the gastrointestinal tract, 95 to 99% of the arsenic is located in erythrocytes, bound to the globin of hemoglobin and is then transported to the other parts of the body. About 70% of the arsenic is excreted mainly through urine. Most arsenic absorbed into the body is converted by the liver to less toxic methylated form that is efficiently excreted in the urine (WHO, 1981; ATSDR, 1989).
Arsenic is a multi-site carcinogen in humans, causing tumors in a variety of tissues including lung, skin, liver, kidney, and bladder, as well as uterus and prostate (NRC, 1999; Waalkes et al., 2000, 2003). Acute arsenic exposure may cause gastrointestinal tract disorders (Goebl et al., 1990), whereas chronic exposure may exert degenerative, inflammatory, and neoplastic changes of the respiratory, hematopoietic, cardiovascular, and nervous systems (Naiger & Osweiler, 1989). Although epidemiological data have firmly established arsenic to be a human carcinogen, animal studies are less well defined. Most laboratory animals appear to be substantially less susceptible to arsenic than humans (Naqvi et al., 1994; Huff et al., 2000). It has been reported that chronic oral exposure to inorganic arsenic (0.05-0.1 mg/kg/day) causes neurological and hematological toxicity in humans but not in monkeys, dogs, and rats exposed to arsenite or arsenate at doses of 0.72 to 2.8 mg/kg/day (Byron et al., 1967).
BIOCHEMICAL METABOLISM OF ARSENICALS
Exposure of humans to inorganic arsenic results in the urinary excretion of inorganic arsenic and two major methylated metabolites, methyl arsenic and dimethyl arsenic (Crecelius, 1977; Smith et al., 1977; Yamauchi & Yamamura, 1979). In many organisms, including humans, inorganic arsenic is reduced in the blood from pentavalent arsenate (As5+) to trivalent arsenite (As3+), then taken up by hepatocytes and oxidatively methylated to monomethyl arsenic acid (MMA) and then to dimethyl arsenic acid (DMA) (Fig. 1) (Aposhian, 1997; Healy et al., 1998; Pott et al., 2001; Thomas et al., 2001). The reduction is facilitated by reductases and the reduced form of glutathione (GSH) along with possibly other thiols as electron donors. Oxidative methylation is carried out by methyltransferases, and S-adenosylme-thionine (SAM) serves as the main methyl donor (Drobna et al., 2005).
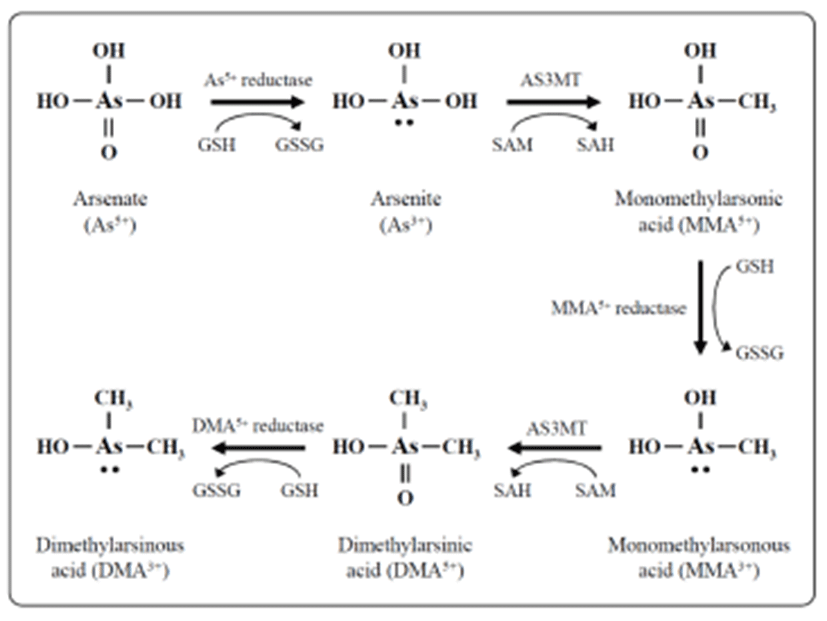
Inorganic arsenic in water is abundant in the form of arsenate; it is negatively charged at physiological pH and slowly taken up by cells (Cohen et al., 2006). Arsenate is rapidly converted to arsenite in vivo (Vahter and Envall, 1983; Vahter & Marafente, 1985) which is taken up by cells much more quickly than arsenate (Tseng, 2009). Methylation of arsenicals facilitates their excretion from the cell and therefore was long considered a detoxification process, but recent evidence indicates that monomethylated and dimethylated arsenicals have many cyto- and genotoxic effects including increased oxidative stress (Kligerman & Tennant, 2007), chromosomal aberrations, and oxidative DNA damage (Schwerdtle et al., 2003; Dopp et al., 2004, 2005). Methylated trivalent metabolites are highly reactive and are more potent inhibitors of GSH reductase and thioredoxin reductase compared with arsenite or pentavalent metabolites (Styblo et al., 1997; Lin et al., 1999). Thioredoxin reductases catalyzes the NADPH-dependent reduction of the disulfide bond in oxidized thioredoxin, which is a critical antioxidant enzyme controlling the cellular redox balance (Rossman, 2003).
More importantly, arsenic can pass through the placenta to the developing fetus. In experimental animals, prenatal exposure to inorganic arsenic has resulted in arsenic accumulation in fetuses, including fetal brain (Rodriguez et al., 2002; Miyazaki et al., 2005), and detection of monomethyl and dimethyl arsenicals in fetuses (Hall et al., 2007). In humans, dimethylarsinic acid (DMA5+) accounts for nearly 90% of all detected arsenic in the blood plasma of both the newborns and their mothers (Concha et al., 1998). This form of arsenic accounts for 60 to 70% of the total detected arsenic in urine of the general population, whereas urine from pregnant women contained more than 90% arsenic as DMA5+. This suggested that arsenic methylation may be increased during pregnancy and that DMA5+ is the major form of arsenic transferred to the fetus.
DEVELOPMENTAL TOXICITY OF ARSENIC
The elevated evidence now suggests that arsenic exposure in utero also poses health risks to the developing fetus (Vahter, 2009). Various epidemiological studies have found significant associations between prenatal arsenic exposure and adverse infant outcomes, such as infant mortality, low birth weight, and birth defects (Ahmad et al., 2001; Milton et al., 2005; Rahman et al., 2007). These health problems were most evident in individuals exposed to high-level arsenic (IARC, 2004). During pregnancy, high-level exposure to arsenic in drinking water causes pregnancy complications, including fetal loss and premature delivery (Chakraborti et al., 2003), and low-level exposure to arsenic affects uterus and placental growth results in progeny birth weight (Hopenhayn et al., 2003; Rahman et al., 2009). Conclusions from these epidemiological studies are further supported by results from animal models (Ferm and Carpenter, 1968; Hood and Bishop, 1972). Several cross-sectional studies have been reported that woman exposure of chronic arsenic in drinking water increases adverse pregnancy outcomes including premature delivery, spontaneous abortion, stillbirth and neonatal death in Bangladesh (Milton et al., 2005), northeastern Taiwan (Yang et al., 2003), and Chile (Hopenhayn-Rich et al., 2000). Despite the strong association between arsenic exposure and a range of child health concerns, the mechanisms by which arsenic elicits these effects remain elusive (Fry et al., 2007; Vahter, 2009; Ahmed et al., 2011).
Arsenic is a well-known teratogen and developmental toxicant in many animal models. Various animal studies have shown that arsenic can produce developmental toxicity, including malformation, growth retardation and fetal death (Tabocova et al., 1996; Golub et al., 1998). Furthermore, developmental toxicity critically depends on dose, route and gestation day following the arsenic exposure (Golub et al., 1998). Fetal malformations were only reported when pregnant rats and mice were intravenous (i.v.) and intraperitoneal (i.p.) injected with inorganic arsenic at early gestation (Stump et al., 1999; DeSesso, 2001). Maternal inhalation or oral ingestion of inorganic arsenic affected fetal development and behavior, but did not cause malformations (Holson et al., 1999; Stump et al., 1999; DeSesso, 2001; Chattopadhyay et al., 2002). The maternal circulation levels of arsenicals are influenced by absorption rates. In oral exposure, arsenic is absorbed into the blood from intestines. It is then transported to the liver and may undergo first-pass metabolism prior to being delivered to the uterus. Intraperitoneal injections, on the other hand, allow arsenic to be taken up by blood vessels directly and may bypass first-pass metabolism (Stump et al., 1999; DeSesso, 2001). In pregnant rats, the maternal blood and embryonic arsenic concentration from i.p. injected mother was significantly higher than those from the orally exposed mother (Hood et al., 1987; Holson et al., 2000a). Inhalation is the least effective means of increasing maternal or embryonic arsenic concentrations, compared to i.p. and i.v. injections and oral exposure (Holson et al., 2000a). Intraperitoneal injection of 45 mg/kg sodium arsenate in Swiss mice during gestation day (GD) 8 induced fetal renal and skeletal malformations without affecting maternal weights (Fascineli et al., 2002). However, oral exposure to arsenic did not cause neural tube defects, even at maternally toxic dose levels. Oral gavage with high-dose arsenic trioxide (10 mg/kg/day) in female Crl:CD(SD)BR rats during GD 14 to 19 decreased fetal weights, but did not induce changes in mating index, fertility index, implantation, or fetal malformation (Holson et al., 2000b). Nevertheless, dimethylarsinic acid (DMA5+), an arsenic metabolite, given to pregnant mice and rats by oral gavage caused developmental toxicity (Rogers et al., 1981; Chernoff et al., 1990). CD-1 mice were orally gavaged with DMA5+ at 200~600 mg/kg/day during GD 7 to 16. These mice showed lower maternal weight gain and fetal weight at 200 mg/kg/day, and a higher incidence of cleft palate at 400 mg/kg/day. Similarly, during GD 7 to 16, CD rats were orally gavaged with DMA5+ at 7.5 to 60 mg/kg/day (Rogers et al., 1981). Maternal weight gain and fetal weight were decreased at >40 mg/kg/day. Fetal mortality was increased at 50 or 60 mg/kg/day, but no fetal gross malformations were seen in these rats. In a later study, DMA5+ was given to pregnant Sprague-Dawley (SD) rats at 40 mg/kg/day by oral gavage during GD 6 to 15. This treatment did not induce maternal weight reduction or maternal lethality, but decreased fetal weight (Chernoff et al., 1990). Furthermore, postnatal developmental changes were observed when arsenic was given in the drinking water to pregnant and lactating rats and continually to the newborns (Rodriguez et al. 2002). Ingestion of drinking water with sodium arsenite (36.7 mg/L) in SD rats during GD 15 to postnatal day (PND) 16 induced developmental indices including delayed pinna detachement on PND 12 and early eye opening on PND 14 in more litters, compared to the untreated controls (Rodriguez et al. 2002). In physiological development, pinna detachment occurs between PND 3 and 4, and eye opening is observed on PND 16. These data indicate that arsenic induces an asynchrony of the maturational processes during postnatal development. Overall, it is assumed that prenatal exposure to arsenic may cause adverse pregnancy outcomes and children’s health problem, however, the exact mechanism underlying arsenic-induced developmental toxicity and a direct relationship with reproductive toxicity remains to be elucidated.
REPRODUCTIVE TOXICITY OF ARSENIC
In males, arsenic may induce gonad dysfunction through declined testosterone synthesis, apoptosis and necrosis (Davila-Esqueda et al., 2012; Shen et al., 2013). However, the male reproductive dysfunction of arsenic exposure on human health is not well established. Recently, few epidemiologic studies have shown that arsenic exposure significantly causes infertility and low sperm quality, as well as erectile dysfunction in men (Nie et al., 2006; Hsieh et al., 2008; Meeker et al., 2010).
Several experimental studies have been demonstrated that significant accumulation of arsenic in testes and accessory sex organs, such as epididymis, seminal vesicle, and prostate gland (Danielsson et al., 1984; Pant et al., 2001). Male mice exposed to sodium arsenite at 20 or 40 mg/L for 5 weeks in drinking water showed decreased epididymal sperm counts and testicular weight (Chang et al., 2007). Similarly, in male rats, daily exposure to sodium arsenite at 5 mg/L for 4 weeks in drinking water also resulted in decreased testicular weights, accessory sex organ weights, and epididymal sperm counts, as well as extensive degeneration of a wide variety of germ cells at stage VII of the spematogenic cycle (Jana et al., 2006). Furthermore, arsenic-exposed mice exhibited dose-dependent gradual reductions in seminiferous tubular diameter and various gametogenic cell populations, such as resting spermatocyte, pachytene spermatocyte, and elongated spermatid (Sanghamitra et al., 2008). These results indicated that arsenic exposure may inhibit the spermatogenesis and sperm development.
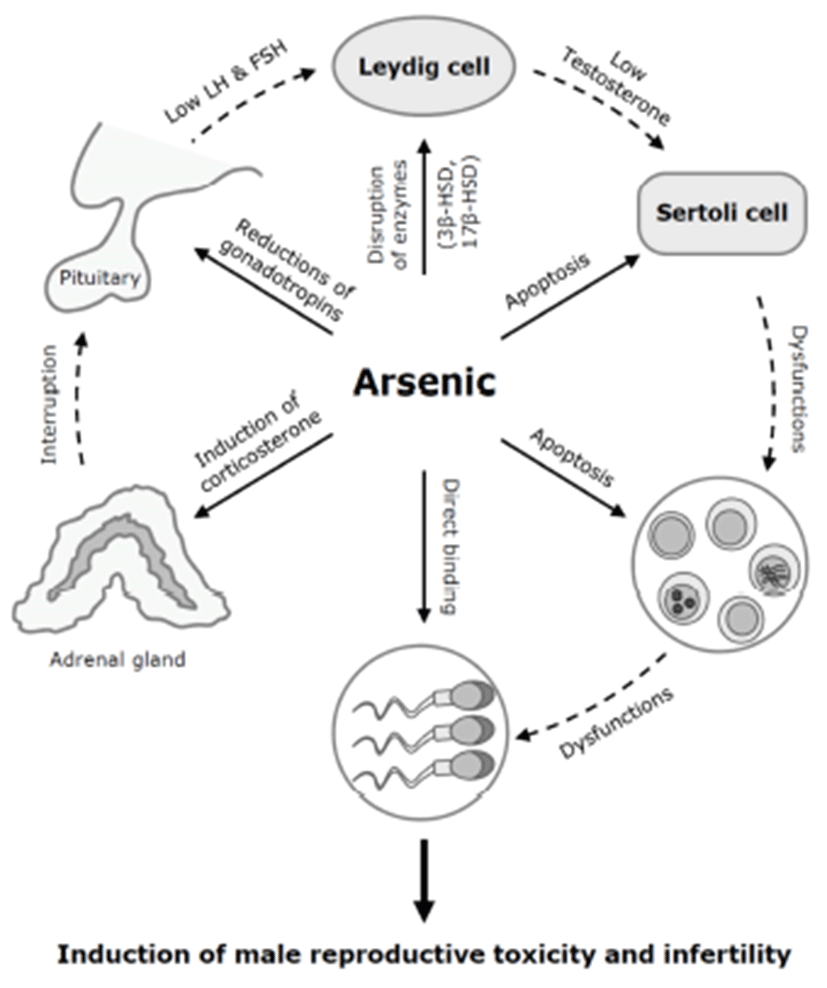
In addition, arsenic exposure also reduced plasma levels of testosterone and gonadotropin in male mice and rats (Sarkar et al.. 2003; Chinoy et al., 2004; Pant et al.. 2004). Sodium arsenite was given to Wister rat via i.p. injections at 4, 5, or 6 mg/kg/day for 26 days. At 5 and 6 mg/kg/day, relative testicular weight, accessory sex organ weights and epididymal sperm counts were decreased. Moreover, sodium arsenite-treated rats were accompanied by decreases of plasma concentrations of luteinizing hormone (LH), folliclestimulating hormone (FSH), and testosterone after arsenic exposure. These suggested that arsenic may target on the brain and/or pituitary as well as directly on the germ cells. However, the molecular mechanisms underlying arsenicinduced male reproductive dysfunctions are poorly understood.
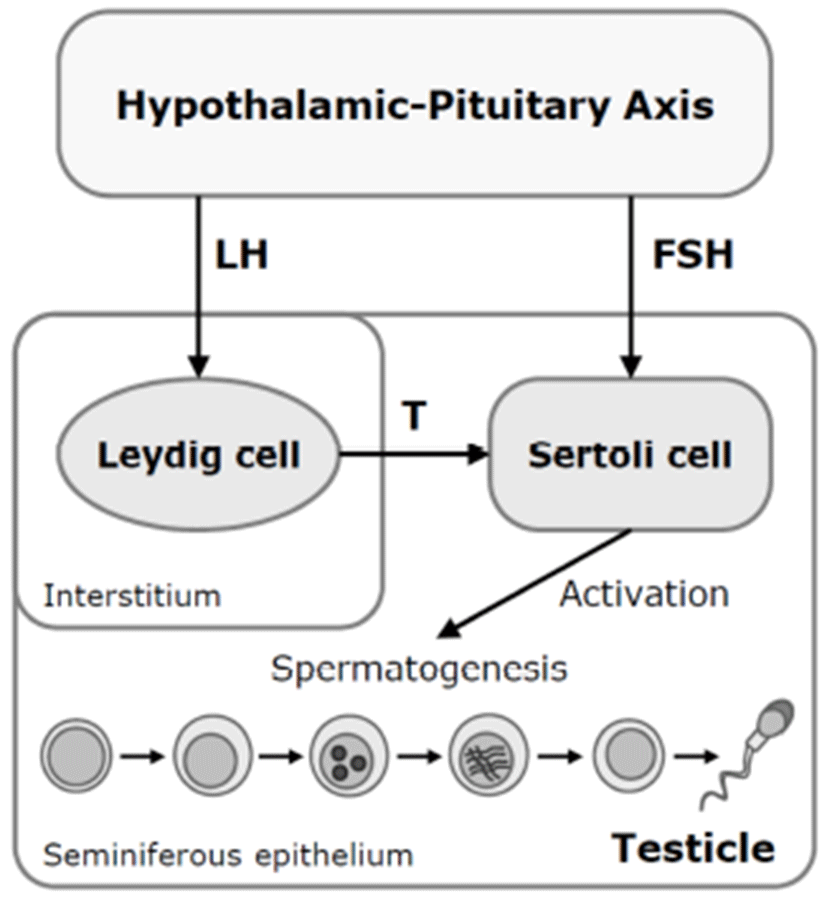
There are several possible mechanisms for the antigonadal activities of toxic chemicals. They might exert a direct inhibitory action on the testis, or they might affect the hypothalamic-pituitary axis causing changes in plasma concentrations of LH and FSH. Reduction of plasma LH might impair Leydig cell function and result in a consequent reduction in testosterone production. Testosterone is a prerequisite for normal spermatogenesis, while FSH is also required for normal testicular function and spermatogenesis (Fig. 2) (Jana et al., 2006). On the other hand, a possible mechanism for decreased sperm motility might be associated with a direct binding of arsenic to sperm (Uckun et al., 2002). Nuclear chromatins of sperm have large amounts of thiol-rich protamines, and the sperm flagellum is rich in thiols. The thiol groups in sperm nuclear or flagellum provide binding sites for arsenic. Another possible cause of the reduction in serum LH, FSH, and testosterone levels could be high serum corticosterone levels. High corticosterone can reduce serum gonadotrophins and testosterone levels (Vreeburg et al. 1988; Hardy et al. 2005), and has been reported in sodium arsenite-treated rats (Biswas et al., 1994).
In addition to spermatogenesis, in mice, cholesterol metabolism and testosterone production were affected by arsenic exposure (Chinoy et al., 2004). Male mice orally exposed to arsenic trioxide (As2O3) at 0.5 mg/kg for 30 days showed increased cholesterol levels and decreased expression of 3β-hydroxysteroid dehydrogenase (HSD) and 17β-HSD, two important regulatory enzyme of steroidogenesis. In the testis, cholesterol is a precursor for testosterone synthesis (Kabbaj et al., 2003). 3β-HSD mainly converts pregnenolone to progesterone, and 17β-HSD converts androstenedione into testosterone. In the seminiferous tubules in the testis, cholesterol in the membrane of developing cells influences the gamete’s fertility (Kabbaj et al., 2003). These data suggest that low plasma level of testosterone after arsenic exposure may be due to low enzymatic conversion by the decreases of 3β-HSD and 17β-HSD, rather than a lack of the synthetic precursor cholesterol.
Arsenic exposure may induce cell death or apoptosis in the testicular germ cells or somatic Sertoli cells in vitro (Celino et al., 2009; Kim et al., 2011). In Japanese eel, low-dose (0.1~1 μM) of arsenic inhibits spermatogenesis via steroidogenesis suppression, while high-dose (100 μM) of arsenic induces oxidative stress-mediated germ cell apoptosis (Celino et al., 2009). We previously showed that arsenic trioxide (As2O3) at 10 μM efficiently induces reactive oxygen species (ROS)-related cytotoxicity and apoptotic cell death in mouse TM4 Sertoli cells (Kim et al., 2011). The testicular Sertoli cells interact directly with developing germ cells throughout the process of spermatogenesis. These cells have an indispensable able in the development and movement of germ cells (Russell, 1993; Griswold, 1998). Thus, spermatogenic dysfunction of arsenic toxicity may be associated with direct damages of testicular component cells (Fig. 3).
CONCLUSION
Arsenic is an important environmental toxicant that affects the reproductive and developmental toxicity. These toxic effects are influenced by the forms, sources, and routes, as well as doses and periods of arsenic exposure. In vivo studies demonstrated that inorganic arsenic, such as sodium arsenite, arsenic trioxide, and dimethyl arsinic acid, an arsenic metabolite, causes reproductive and developmental toxicity. Prenatal exposure to inorganic arsenic by only i.p. or i.v. injection routes causes fetal malformation; in contrast, oral and inhalational exposure to inorganic arsenic affects fetal development, including growth retardation and fetal death. In drinking water, oral exposure to inorganic arsenic causes dysfunctions of spermatogenesis, reductions of testosterone and gonadotrophins, and disruptions of steroidogenesis. However, the reproductive and developmental toxicity of arsenic is poorly understood, and the molecular mechanism of arsenic-induced reproductive toxicity remains unclear. Therefore, we further investigated some of the possible mechanisms that are affected by arsenic causing reproductive toxicity. The crucial mechanisms of arsenic-induced reproductive toxicity may be associated with hormonal regulation and function, binding to sperm, and regulation of steroidogenesis, as well as direct effects of testicular component cells.