





INTRODUCTION
Mesenchymal stem cells (MSCs) are multipotent stem cells that are capable of self-renewal, proliferation, and differentiation into multi-lineage cell types (Galderisi & Giordano, 2014). They also exhibit the unique immunosuppressive properties by modulating the proliferation and functions of the immune cell populations (Perico et al., 2013). Thus, MSCs are expected to replenish the cellular component of damaged host tissue without disrupting the local microenvironment after transplantation. Currently, MSCs transplantation has been proposed as a promising therapeutic strategy for various degenerative diseases such as liver and heart (Nhung et al., 2015, Montanari et al., 2015). MSCs are isolated from the various locations in the body including bone marrow, skin, hair follicles, dental pulp, adipose tissue, umbilical cord, amniotic membrane, endometrium, placenta and synovium (Li et al., 2014, Beltrami et al., 2006). Among MSCs, human adipose-derived stem cells (hADSCs) seem to be suitable for the clinical application due by high stem cells yield from lipoaspirates, faster cell proliferation, and less discomfort during harvesting procedure (Liao & Chen, 2014).
One important problem in the cell-based therapies is the delivery of the cells to the site of injury, a process termed ‘homing’. The therapeutic efficacy of MSCs is greatly dependent on their ability of migration to the diseased sites, which is influenced by multiple factors including age and passage number of the cells, culture conditions, and the delivery method (Potapova et al., 2007, Kavanagh et al., 2015). The migration and homing of MSCs to the tissue of injury is influenced by various growth factors and cytokines. The interactions of stromal cell-derivedfactor-1α (SDF-1α) and C-X-C chemokine receptor type 4 (CXCR4) are very important for the migration of transplanted MSCs (Karp & Leng Teo, 2009). Thus, stimulation with multiple cytokines such as SDF and HGF can up-regulate CXCR4 expression of MSCs and increase in vitro migration capacity to SDF-1 (Shi et al., 2007). Additionally, Plateletderived growth factor (PDGF) is known to act as a potent stimulus of cell migration. During embryonic blood vessel formation in the mouse, angiogenic sprouting and vessel enlargement were shown to involve co-migration and proliferation of vascular smooth muscle cell/pericytes progenitor in a PDGF-B-dependent manner (Hellström et al., 1999). Human choroid fibroblasts were elongated and migrated in response to three isoforms of PDGF including AA, AB, and BB types in an in vitro wound assay (Nagineni et al., 2005). Human endometrial stromal cells have been shown to migrate by PDGF-BB stimulation through the activation of both extracellular signalregulated kinase 1/2 (ERK1/2) and whether phosphatidylinositol 3-kinase (PI3K)/Akt signaling pathways (Gentilini et al., 2007). Human adult stem cells such as hADSCs and bone marrow-derived MSCs (BM-MSCs) also exhibited PDGF-AB-dependent high migration activity (Ponte et al., 2007). In human and rabbit MSCs, either PDGF-AB or PDGF-BB could induce both migration and proliferation of MSCs in a microchemotaxis chamber (Ozaki et al., 2006). These observations suggest that PDGF could play a pivotal role to enhance the homing of MSCs into the target tissues after transplantation of MSCs. Fibroblasts in medium containing PDGF were observed to migrate as individuals (Rhee et al., 2009).
Matrix metalloproteinases (MMP) are a family of zincdependent proteolytic, major function of them are degraded various components of the extracellular matrix (ECM) and mediate ECM remodeling in biological process. Under physiological conditions, MMP activities are regulated at multiple levels such as gene expression, activation of zymogens and interaction with specific inhibitors in order to limit MMP activity (Chen et al., 2013, Raffetto & Khalil, 2007). Moreover, MMP promote recruitment of stem/progenitor cells and facilitate migration of MSCs. Human adult olfactory stem cells exhibited different patterns of expression for MMP1, MMP2, MMP9, and MT1-MMP upon cell migration when compared with non-migrating cells (Ould-Yahoui et al., 2013). Furthermore, BM-MSCs has been shown at least partially regulated by MMP2 and high culture confluence decreased transendothelial migration of MSC with an increased production of the natural MMP inhibitor TIMP-3 (De Becker et al., 2007). MMP1 activates protease activated receptor (PAR)-1 to induce cell invasion, motility and angiogenesis (Gehmert et al., 2010, Kim et al., 2013).
This study focused on the mechanism whereby PDGF-BB could induce the migration of hADSCs in vitro. Particularly, molecules involved in signaling pathways such as PI3K, ERK, and myosin light chain kinase (MLCK), and role of specific MMP molecules have been investigated.
Materials and Methods
The human abdominal adipose tissue was obtained from six patients undergoing liposuction with informed consent in local hospitals in Korea. All experiments were approved by Institutional Review Board of Seoul Women’s University. Red blood cells in adipose tissue samples were removed by several washing step with Dulbecco’s phosphate-buffer saline (DPBS; Gibco). Then, tissue was mixed with 1x volume of 0.15% type I collagenase (Gibco) and incubated at 37℃ for 1 hour with gentle shaking. After that, 1X volume of DMEM (5.5 mM; DMEM-LG; Gibco) containing 10% FBS (Gibco), 100 U/mL penicillin, 0.1 mg/mL streptomycin, and 3.7 mg/mL sodium bicarbonate was added and centrifuged at 3,000 rpm for 10 min. Cell pellets were washed, and then cells were plated in 25 cm2 culture flask containing 5 mL of the same medium. Cells were cultivated at 37℃ with 5% CO2. When adherent cells were 70~80% confluent, cells were detached using 0.125% trypsin and 1 mM EDTA solution at 37℃ for 2 min. These cells were stored at –190℃ in a nitrogen tank until use. After thawing, cryopreserved cells were examined for their stem cell properties, expansion ability and differentiation potential for three mesodermal lineages as described previously (Kang et al., 2011). These cells were named as hADSCs and used.
For the experiments, frozen-thawed cells were resuspended in the same culture medium at a density of 4,000 cells/cm2. The culture medium was changed twice a week. hADSCs between passage (p) 3 and p7 were used throughout this study. Of six cell lines established, different cell line was used each experiment, and data were obtained from three or more cell lines. PDGF-BB was purchased from Peprotech (#100-14B, Korea), and dissolved in 0.1% BSA solution. It was frozen in aliquots in 0.7 mL tube and stored at –20℃.
hADSCs were seeded in 12-well at a density of 2.8×104 cells/cm2 and cultured with DMEM containing 10% FBS. After overnight, the monolayer cells were scratched manually with a 1-mL pipette tip, and after two washes with DMEM. Cells were cultivated with DMEM containing 4% of bovine serum albumin (BSA) and 50 ng/mL PDGF-BB. Photographs of wound area were taken at time 24 and 48 hours. Using the Image J software, the area of each wound was calculated at each time point.
Cell migration was determined using transwell chambers in which two chambers were separated by a polycarbonate membrane (pore size, 8.0 μm; diameter, 6.5 mm). Total 1×105 cells/100 μL were suspended within transwell inserts (upper chamber) in DMEM containing 0.5% BSA. Wells of 24-well dish (bottom chamber) were filled with 650 μL of the same medium containing 50 ng/mL PDGF-BB with or without BB94 (Batimastat, Tocris, #2961), ARP100 (Santa Cruz, #sc-203522), LY294002 (Cell signaling, #9901), or Y27632 (Sigma Aldrich, #Y0503). After incubation for 2 days at 37℃ with 5% CO2, cells that migrated to the lower chamber were counted. Results were obtained from three independent experiments.
Total RNA was isolated using Tri-reagent (Ambion, #15596-026) according to the manufacturer’s instructions. Total 7.5 µg of RNA was reverse-transcribed using the following RT mixture: 20 mM MgCl2 (Bio Basic), 10× PCR buffer (Bio Basic), 10 mM dNTPs mixture (Bio Basic), 0.5 mg/mL oligo (d)T15 (Bionics) for RT-PCR or 0.5 mg/mL Random hexamer for qRT-PCR, 40 U/μL RNase inhibitor (Bio Basic) and 200 U/µL AMV-RT (Invitrogen, #28025-013). RT reaction was performed for 60 min at 37℃, and PCR reaction was carried out using the following PCR mixture: 25 mM MgCl2, 10× PCR buffer, 5U/μL Taq polymerase, 2.5 mM dNTPs, 10 µM forward and reverse primers. Amplification was performed for 25 or 35 cycles at a denaturing temperature of 94℃ for 30 sec and an extension temperature of 72℃ for 30 sec. Annealing temperature was set depending on the species of primer. The PCR products were mixed with 6× loading buffer (0.25% bromophenol blue, 0.25% xylene cyanol and 40% sucrose) and separated on 3% agarose gels. After electrophoresis, gels were stained with ethidium bromide. DNA signals on the gels were imaged under UV light using an image analysis system (ULTima, Hoefer) qRT-PCR was performed in total volume 20 μL buffer solution containing 1 μL of template cDNA, 10 μL SYBR Green I Master (Roche, #04707516001), and 10 pM of each primer using Light Cycler 480 Real-Time System (Roche). Relative expression levels of cDNA were normalized to the expression of 18s rRNA using the comparative CT (2-∆∆CT) method. Primers used in this study were shown in Supplementary Table S1. All PCR mixture components were purchased from Takara (#R011).
All samples were homogenized in a lysis buffer consisting of 50 mM Tris-HCl (pH 8.0), 150 mM NaCl, 1% sodium deoxycholate, 1% Triton X-100, 1% sodium dodecyl sulfate (SDS), a protease inhibitor (Roche, #04693116001) and a phosphatase inhibitor (Sigma Aldrich, #P0044) for 30 min on ice. Lysates were clarified by centrifugation at 13,000 rpm for 20 min at 4℃. Thirty μg of protein was mixed with 2X sample buffer consisting of 12.5 mM Tris-HCl (pH 6.8), 4% SDS, 10% 2-mercaptoethanol, 20% glycerol, 0.004% bromophenol blue and then heated at 70℃ for 5 min. The protein samples were separated on 10% SDS polyacrylamide gel electrophoresis (SDS-PAGE) gels and the proteins were then transferred to nitrocellulose membranes. After blocking in 3% BSA for 1 hour, the membrane was incubated with a mouse monoclonal antibody to MMP1 (1:1,000, Santa Cruz, #sc-21731), rabbit polyclonal antibody to MMP2 (1:1,000, Abcam, #ab37150), rabbit monoclonal antibody to phosphorylated extracellular signal-regulated kinase (ERK; 1:1,000, Cell signaling, #4377), rabbit polyclonal antibody to ERK (1:1,000, Cell signaling, #9102), rabbit polyclonal antibody to phosphorylated (p)-myosin light chain (MLC; 1:1,000, Cell signaling, #3674), rabbit polyclonal antibody to MLC (1:1,000, Cell signaling, #3672) or mouse monoclonal antibody against β-actin (1:1,000, Santa Cruz, #sc-47778) in a TBST blocking buffer consisting of 150 mM NaCl, 0.1% Tween-20, 20 mM Tris (pH 7.5) and 3% BSA for overnight. After washing, the membrane was incubated in HRP-conjugated, secondary goat anti-mouse IgG (1:5,000, Santa Cruz, #sc-2005), or HRP-conjugated, secondary goat anti-rabbit IgG (1:5,000, Santa Cruz, #sc-2004) in a blocking buffer for 1 hour. The stained membranes were visualized by ECL kit (Thermo, #32109).
hADSCs were cultivated in DMEM containing 10% FBS in 6-well at a density of 1×104 cells/cm2. siRNA duplex (Bioneer) or Lipofectamine RNAiMAX (Invitrogen, #13778- 150) complex was dissolved in 250 μL DMEM, and incubated for 5 min. Then, dissolved siRNA duplex and Lipofectamine were mixed and incubated for 15 min. After adding 1.5 ml DMEM to the 500 μL mixture, the diluent was added to the cells and incubated for 4 hour at 37℃ with 5% CO2. Following washing cells with a fresh DMEM containing 10% FBS, cells were incubated for overnight. siRNA used in this study are shown in Supplementary Table S2. Predesigned siRNAs for the control, MMP1 and MMP2 molecules were provided by Bioneer.
| Refseq accession number | Gene symbol | Sense siRNA sequence | Antisense siRNA sequence |
|---|---|---|---|
| NM_002421.3 | MMP1 | GAAAUCUUGCUCAUGCUUU | AAAGCAUGAGCAAGAUUUC |
| NM_004530.5 | MMP2 | CUGCAAACAGGACAUUGUA | UACAAUGUCCUGUUUGCAG |
Caseinase and gelatinase activities in hADSCs-conditioned medium was examined using zymography. Ten microliter of the conditioned medium was mixed with the same volume of non-reducing sample buffer consisting of 62.5 mM Tris-HCl, 25% glycerol, 4% SDS, and 0.01% bromophenol blue (pH 6.8). Casein and gelatin substrate for SDS-PAGE were prepared by adding 1 mg/mL β-casein or bovine skin type B gelatin to the 8% resolving gel. After electrophoresis, casein-and gelatin-SDS gels were soaked with 2.5% Triton X 100 in 50 mM Tris-HCl buffer (pH 8.0) for 30 min with gentle stirring. Then, gels were washed in a developing buffer (5 mM CaCl2, 0.02% NaN3, 50 mM Tris-HCl, pH 8.0) and incubated in fresh incubation buffer for overnight at 37℃ to develop the enzyme activity. At the end of the incubation, the gels were stained with a solution of coomassie brilliant blue R-250 in 27% methanol for 30 min, followed by destaining with 10% acetic acid in 25% methanol. Clear bands against the blue background indicated the presence of degradative activity of MMPs.
Results
To investigate the effect of PDGF-BB on hADSCs migration during in vitro culture, cells were cultivated in the presence of 0, 2, 10, or 50 ng/mL of PDGF-BB and the migration rates were measured using transwell assay. Number of migrated cells was 22.7±4.7, 71±5.7, 120.7± 14.7, and 164.0±32.6 at 0, 2, 10 or 50 ng/mL, respectively, (Fig. 1A). The results demonstrate that PDGF could enhance migration of hADSCs in a dose-dependent manner. The enhanced migration by PDGF treatment, however, was not due to increased cell proliferation by the PDGF treatment since little difference was observed in total cell number among different PDGF treatment groups (Fig. 1B). In the presence of 50 ng/mL of PDGF, more cells migrated over wound area compared to the cells in the absence of PDGF (Fig. 1C). When measured, the percentage of migrated area was increased 22.8%±2.9 after 1 day, and 19.5% ±5.1 after 2 days (Fig. 1D, p<0.05). The results demonstrate that increased number of migrated cells could enhance the wound healing.
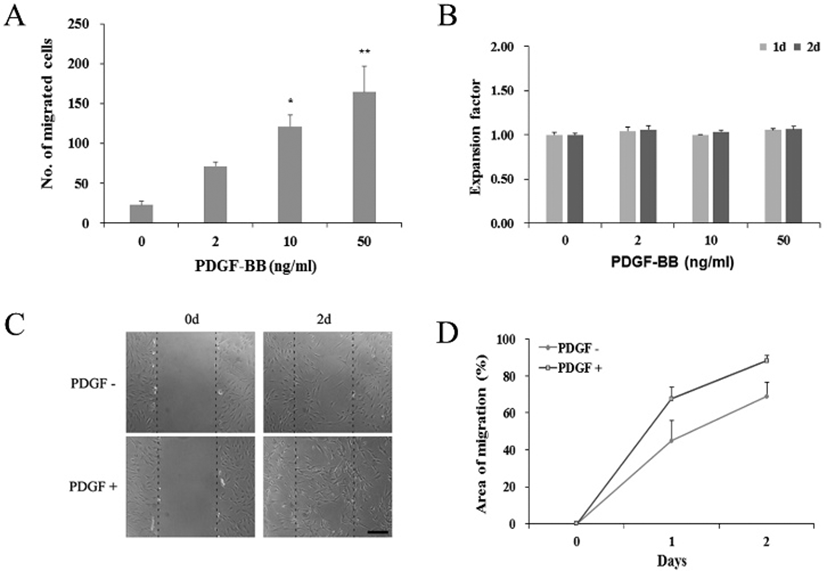
To see if the observed PDGF effects might have occurred via the PDGF receptor signaling, we examined the expression of PDGF receptor genes in PDGF-treated hADSCs. qRT-PCR results showed that, regardless of the 50 ng/mL PDGF treatment or not, cells constantly expressed both PDGFRα and PDGFRβ genes (Fig. 2). These results suggest that PDGF effect might take place via its receptor on the surface of the hADSCs.
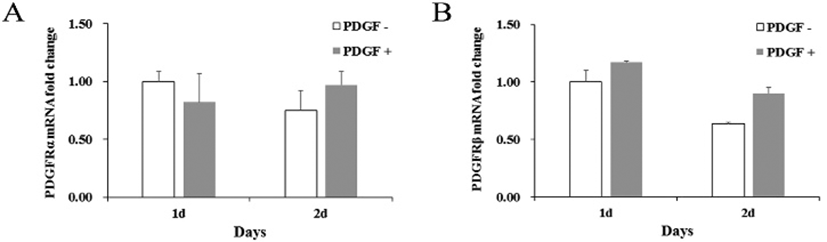
If MMP molecule could be involved in the PDGF-induced migration, effect of MMP inhibitors was examined on the migration. A general MMP inhibitor, 10 μM BB94, reduced the migration to 36.9% ±10.0 and a MMP2 inhibitor, 20 μM ARP100, reduced to 29.1% ±3.5 compared to PDGF alone-treated group (Fig. 3). These results demonstrate that MMPs might play an important role in the PDGF-induced migration process. To address a role of specific MMPs in PDGF-induced migration, expression of various MMP genes was examined. qRT-PCR results showed that genes of MMP2, MMP3, MMP7 and MMP9 were consistently expressed and the level of expression did not change significantly regardless of the PDGF treatment. In contrast, PDGF treatment greatly increased MMP1 gene expression by 9.5±2.6-fold after 1 day, and by 11.5±5.9-fold after 2 days, compared to the control (Fig. 3B). These results suggest that MMP1 could play a role in the migration.
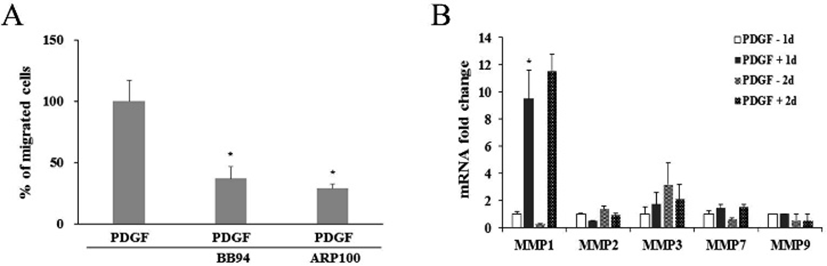
It was further examined whether PDGF or MMP inhibitors might influence on the protein expression and enzyme activity of MMP1 and MMP2. When cells were treated with PDGF alone or with a MMP inhibitor, western blot results showed little change in comparison to the untreated control (Fig. 4A). However, treatment of either BB94 or ARP100 during zymography mostly inhibited the caseinolytic activity of MMP1 or gelatinolytic activity of MMP2. These results suggest that inhibition of PDGF-induced cell migration by BB94 or ARP100 might occur via inhibition of MMP1 or MMP2 enzymatic activity, but not via the suppression of MMP gene expression (Fig. 4B, 4C).
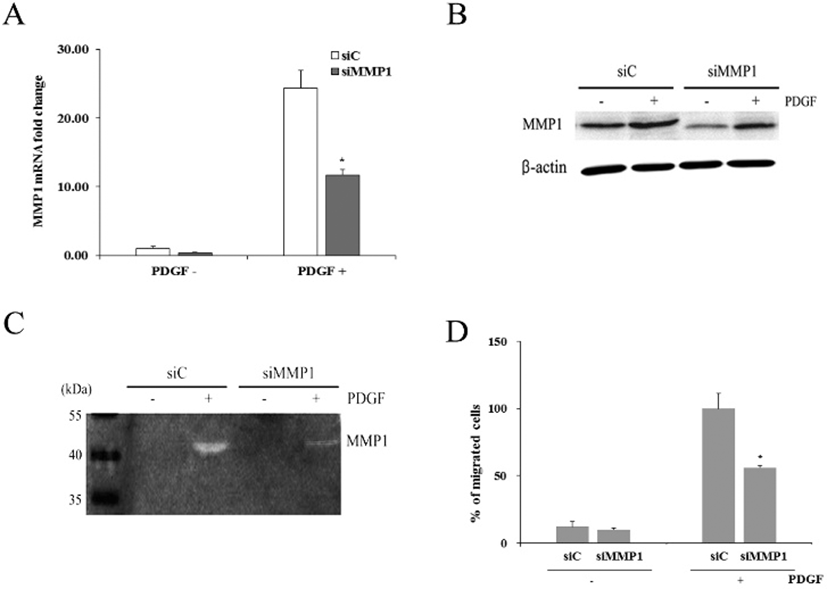
When cells were transfected with siMMP1, they showed reduced expression of MMP1 mRNA by 0.41±0.04-fold (Fig. 5A). When both control and transfected cells were treated with PDGF for 1 day, control cells (siC) showed increased MMP1 expression by 23.7±2.5-fold. Similarly, transfected cells (siMMP1) also showed increased MMP1 expression by 11.4±0.8-fold. However, compared to the MMP1 mRNA expressed in siC, only about 48% of MMP1 mRNA was expressed in siMMP1.
Western blot analyses demonstrated that siC produced distinct amount of MMP1 protein, whereas siMMP1 produced markedly reduced amount of the protein. However, PDGF treatment of both types of cells increased the protein amount, particularly in siMMP1 (Fig. 5B).
Casein zymography revealed that both conditioned media obtained from siC and siMMP1 culture in the absence of 50 ng/mL PDGF showed little enzyme activity of MMP1 (Fig. 5C). In contrast, both media from siC and siMMP1 culture in the presence of PDGF showed distinct caseinolytic activity. However, the enzymatic activity of hADSCs were greatly diminished by siMMP1 transfection.
When cell migration assay was done on hADSCs transfected with siC, percentage of migrated cells was 12.1%± 3.6 (Fig. 5D). hADSCs transfected with siMMP1 showed similar migration rate. When both of these cells were treated with PDGF, siC showed about 100% whereas only 56.2%± 1.7 of siMMP1 migrated to the lower chamber. The results support the above suggestion that MMP1 might play an important role in PDGF-induced hADSCs migration.
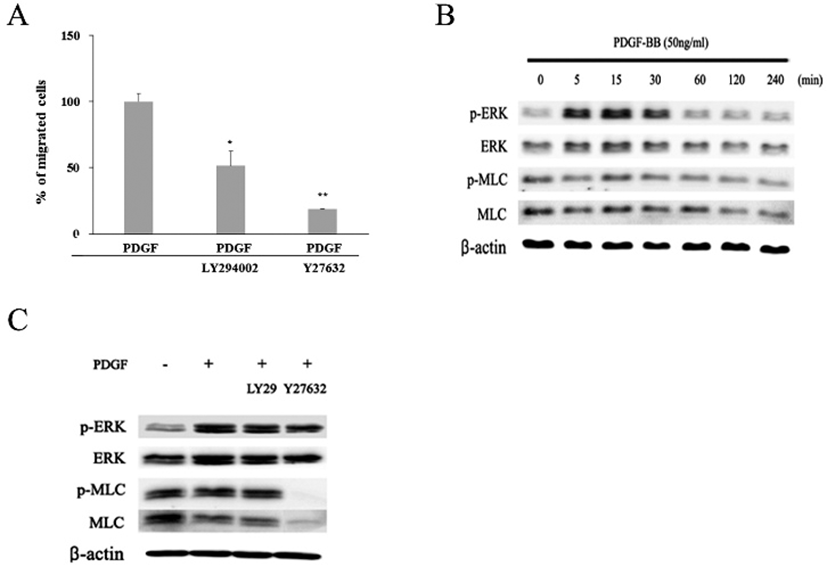
To investigate whether PI3K and/or Rho/ROCK signaling pathway might be involved in the PDGF-induced migration of the hADSCs, effects of LY294002, a PI3K inhibitor, or Y27632, a ROCK inhibitor, on the migration were examined (Fig. 6A). When cells were treated with LY294002 together with PDGF, percentages of migrated cells was diminished to 51.5%±11.2. Moreover, when cells were treated with both Y27632 and PDGF, migration was greatly inhibited to 18.6%±0.4. These results demonstrate that both PI3K and Rho/ROCK pathway could be involved in the PDGF-induced migration.
To verify whether MAP kinase might be involved in the migration, expression of active phosphorylated form of ERK (p-ERK) was examined by western blot. The appearance of one of the key components of cell locomotion, an active phosphorylated MLC (p-MLC), was also examined (Fig. 6B). In the absence of PDGF, cells showed very small amount of p-ERK protein in contrast to a larger amount of ERK, MLC, and p-MLC proteins. When cells treated with PDGF were examined by the same method, they showed greatly increased amount of both ERK and p-ERK proteins. The amount of p-ERK protein markedly increased during 5 to 30 min after PDGF treatment (Fig. 6B). Thereafter, the expression decreased to a similar level as the beginning. In contrast, there was little significant change in the amount of both MLC and p-MLC proteins whether cells were treated with PDGF or not. These results imply that signaling pathway of PDGF-induced migration might involve p-ERK molecule. To further investigate a possible involvement ERK and MLC proteins in the migration, cells were examined at 15 min after treatment with either LY294002 or Y27632 together with PDGF. Again PDGF increased the amount of both ERK and p-ERK proteins (Fig. 6C). However, LY294002 treatment did not induce further changes in the amount of ERK or p-ERK as well as MLC and p-MLC proteins. In contrast, treatment with Y27632, a ROCK inhibitor, almost abolished the appearance of both p-MLC and MLC proteins within 15 min even in the presence of PDGF. These results show that p-MLC molecule plays an important role in the PDGF-induced migration. In addition, the results suggest that the PDGF signaling appears to propagate by two separate pathways, one including PI3K molecule and another not.
Discussion
Present study demonstrates that MMP, particularly MMP1, molecule might play an important role in PDGF-induced migration of hADSCs in vitro, and both PI3K and ROCK pathway could be involved in the migration via MLC activation.
Similar to other types of cells (Fan et al., 2015, Kim et al., 2011), we have also observed that PDGF-BB could induce migration of hADSCs in vitro. As well as fresh hADSCs, frozen-thawed hADSCs which have been used in the present study exhibited a PDGF-induced migration effect in a dose-dependent manner. These cells also expressed both PDGFR-α and -β similar to the previous reports in which both types of receptor were constitutively expressed in most hADSCs regardless of passage number as revealed by immunostaining and RT-PCR methods (Ryu et al., 2013, Hye Kim et al., 2015). PDGF-BB forms both homodimer and heterodimer of PDGFR-α and PDGFR-β, while PDGF-AA bind only PDGFR-α. However, it is well known that PDGF-BB preferentially interacts with PDGFR- β, while it has a lower binding affinity for PDGFR-α (Andrae et al., 2008). Recent work has shown that PDGFR-β is mainly activated by PDGF-BB during MSC migration and it’s signal cascade could be enhanced by fibronectin through the PI3K/Ark and FAK pathways (Veevers-Lowe et al., 2011). Based on these information, PDGF effects seen in frozen-thawed hADSCs appear to occur via PDGFR- β present on the surface of hADSCs.
When cells were treated with PDGF and either one of MMP inhibitors, BB94 or ARP100, we found that both inhibitors markedly inhibited the PDGF-induced migration. BB94 is known to inhibit many MMPs including MMP1, 2, 3, 7, and 9. While ARP100 is more potent against MMP2, it can inhibit other MMPs such as MMP9, and 13 (Rossello et al., 2004). Thus it is suggested that MMP molecule(s) might be involved in the migration. In the subsequent studies in which mRNA levels of various MMPs were quantitatively examined, we observed that PDGF dramatically increased MMP1 gene expression. However, it did not induce any significant changes of MMP2, 3, 7 or 9 expression, which was consistently observed in several different lines of hADSCs. Although western blot results showed only a small increase of MMP1 protein, casein zymogram demonstrated distinct appearance of MMP1 enzymatic activity following PDGF treatment. Moreover, caseinolytic activity of hADSCs were greatly diminished by siMMP1 transfection such that only 56.2% of siMMP1-transfected cells exhibited migration when compared to the control, siC-transfected cells. Importance of MMP1 in cell migration has also been described in other cells. PDGF-BB has been shown to induce MMP1 expression in human dermal fibroblasts (Endo et al., 2003). Human BM-MSCs were shown to consist of several populations, and highly migrating cells among the populations exhibited enhanced MMP1 gene expression and tumor-trophic activity compared with poorly migrating MSCs. Moreover, blocking the interaction of MMP1 and its cognate receptor PAR1 effectively diminished the migratory ability of MSCs, suggesting that MMP1 is critically involved in the migration capacity of BM-MSCs (Ho et al., 2014). Even an in vivo study using MDX mice model with dystrophic skeletal muscles showed that MMP1 treatment could enhance myoblast migration resulting in the faster muscle regeneration (Wang et al., 2009). These observations suggest that MMP1 molecule might play a major role in PDGF-induced migration of hADSCs,
Many investigators have reported about the role of MMP2 in the migration of human cells as well as animal ones. Human hematopoietic stem cells have been suggested to egress from BM by MMP2 which was released from BM-MSCs upon activation by G-CSF (Ponte et al., 2012). Trophoblast cells were inhibited to migrate and invade by upregulation of miR-519d-3p targeting MMP2, and knock-down of MMP2 using a siRNA attenuated the increased trophoblast migration and invasion promoted by the miR-519d-3p inhibitor (Ding et al., 2015). During angiogenesis of the microvascular endothelial cells, hADSCs were shown to release extracellular vesicles containing more proMMP2 and active MMP2 following stimulation with PDGF (Lopatina et al., 2014). We have also observed that PDGF-induced migration of hADSCs was inhibited by ARP100. As ARP100 is known to be a MMP2 inhibitor, MMP2 is likely to be involved in this migration. However, PDGF did not induce MMP2 gene expression in hADSCs. Thus MMP2 enzymatic activation rather than MMP2 protein synthesis appears to be involved.
We have observed that Y27632, a ROCK inhibitor, markedly inhibited the PDGF-induced migration, which was accompanied with a decreased amount of MLC and p-MLC proteins. Y27632 has long been used to inhibit MLC function in various cells. Bovine hyalocytes exhibited a contractile property and maximum phosphorylation of MLC in the presence of PDGF-BB, both of which were inhibited by Y27632 (Hirayama et al., 2004). Migration of rat vascular smooth muscle cells was induced by asymmetric dimethylarginine (ADMA), in which ADMA activated Rho/ROCK signal pathway. Pretreatment with ROCK inhibitor, Y27632, however, completely reversed the induction of ADMA on ROCK and in turn inhibited ADMA-induced cell migration (Zhou et al., 2014). In human airway smooth muscle cells, PDGF-BB-induced cell migration was inhibited by tocotrienol, a ROCK inhibitor (Harada et al., 2015). In the present study, PDGF-induced migration of hADSCs was similarly inhibited by Y27632 accompanied by the inhibition of p-MLC. Based on these information, it is rather apparent that migration of hADSCs by PDGF-PDGFR signaling occurs through the RhoA/ ROCK pathway leading to the phosphorylation of MLC protein.
In an earlier study, treatment of human BM-MSC with a PI3K inhibitor was shown to abolish the PDGF-induced effect, suggesting that PDGF signaling might happen via PI3K pathway (Kratchmarova et al., 2005). Later it was observed that the PDGF-BB effect happened via activation of the PDGFR-β, and the signal cascade was enhanced through the PI3K/Ark and FAK pathways (Veevers-Lowe et al., 2011). In the present study, migration of hADSCs induced by PDGF-BB treatment was inhibited by LY 294002, supporting the previous findings that cell migration by PDGF might occur via the PI3K signaling pathway. However, many findings have also suggested an involvement of ERK activity in PDGF signaling pathway. During PDGF-BB-stimulated basal migration of endometrial stromal cells, either PI3K/Akt inhibitor or ERK1/2 inhibitor exhibited that both signaling pathways were required for the induced cell motility (Gentilini et al., 2007). In response to stromal cell-derived factor, BM-MSCs exhibited increased migration, and the migration was accompanied with phosphorylation of ERK1/2 (Gao et al., 2009). In human airway smooth muscle cells, PDGF could facilitate cell migration through the ERK pathway and ERK inhibitor showed inhibition of migration (Ito et al., 2009). Recently hADSCs have also been shown to migrate via Akt and ERK phosphorylation upon PDGF-D treatment, and the induced migration was inhibited by LY294002 (Hye Kim et al., 2015). While all these information indicate that PI3K-ERK pathway mediates PDGF effect leading to the migration of cells, it does not seem to be the only pathway leading to the ROCK activation. We have seen that PI3K inhibitor diminished the migration by only about 50%, and it induced little change of ERK and p-ERK proteins. In contrast, Y27632, a ROCK inhibitor, inhibited more than 80% of cell migration, and almost abolished the appearance of both MLC and p-MLC proteins within 15 min following PDGF treatment. These observations suggest that PDGF-induced migration of hADSCs might involve other signaling pathway in addition to the PI3K pathway. In other migratory cells, role of MAPK signaling pathway has been suggested in the induction of MMP1 expression. When gastric epithelial AGS-GR cells were treated with gastrin, the expression and enzymatic activity of MMP1 as well as the migration of the cells increased, and the effects have been suggested to occur via protein kinase C and p42/44 MAP kinase (Kumar et al., 2015). Similarly, TNF-α could induced both migration of breast cancer cells and expression of MMP1/3, and these events have been shown to act via p38 MAPK (Xia et al., 2015). Even in nonmigratory fibroblasts, methotrexate treatment was shown to involve ERK1/2 signaling pathway resulting in the MMP1 expression (Nabai et al., 2015). Further studies are needed to clarify if MAPK pathway might be involved in the PDGF-induced MMP1 expression in hADSCs.
Finally, in transwell assays, we observed that less than 0.2% of cells have migrated to the lower chambers. Similar lower numbers of cell migration has been previously observed in BM-MSCs as well as hADSCs (Maijenburg et al., 2009). The work has also shown that the migration abilities of these cells greatly increased responding to the stromal cell-derived factor-1α or PDGF-BB, particularly, in the presence of extracellular matrix. Considering that a possible existence of various other molecules in addition to these around target cells in vivo, actual cell numbers that migrate upon stimuli might vary under circumstances.
In conclusion, we have observed that PDGF treatment markedly increased migration of hADSCs in vitro, accompanying a greater MMP-1 mRNA expression. The PDGF-induced migration was diminished by general MMP inhibitor, and silencing of MMP-1 mRNA markedly reduced the PDGF-induced migration. ROCK inhibitor prevented PDGF-induced migration and appearance of MLC and p-MLC proteins. PI3K inhibitor showed about 50% Inhibition of migration and little changes of ERK and p-ERK. These results suggest that PDGF might signal hADSCs through PI3K, and MMP1 activity could play an important role in this PDGF-induced migration in vitro.