
INTRODUCTION
Colorectal cancer (CRC), also known as colon cancer, is the third common type of cancer in humans with lifetime risk of 5% (Siegel et al., 2017). Surgical resection and subsequent chemotherapy and/or radiotherapy are the traditional treatments for this cancer, but the use of chemotherapeutic drugs often is limited since these drugs are not specific to cancer cells but give severe toxic effects on normal proliferative cells such as intestinal gland cells. Over the last decades, new therapeutic regimens for CRC treatment such as molecular targeted therapy have been employed. However, as seen in almost all other cancers, drug resistance and serious side effects of drugs also occur, which result in the relapse of tumor and cessation of drug administration respectively (Britten, 2013). Thus, it is of clinical significance in the area of oncology to develop new therapeutic strategy to overcome drug resistance and reduce the incidents of side effects.
The PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways play key roles in controlling cell survival, proliferation, differentiation and motility in response to various growth factors (Mendoza et al., 2011). Aberrant regulation induced by gene mutation or amplification in the components of these signaling pathways is implicated in malignant transformation, tumorigenesis and drug resistance (McCubrey et al., 2007). Thus, much effort has been made to develop molecular inhibitors targeting these pathways (De Luca et al., 2012). During the last decade, simultaneous treatment with two pathway inhibitors has been suggested for more effective therapeutic intervention, since cross-talk and pathway convergence between these two pathways have been described and cancer cells with RAS mutation is capable of signaling through both pathways (Mendoza et al., 2011). However, the combined targeting strategy often proved to cause significant unacceptable drug-related toxicity, resulting in termination of clinical trials (Britten, 2013).
Metformin, a biguanide derivative, has been widely prescribed for treatment of type 2 diabetes mellitus for over 50 years and proved a well-tolerable drug with low cost. Several retrospective and case-control studies indicated that type 2 diabetes mellitus is associated with high risk for certain types of cancer including CRC (Larsson et al., 2005; MacKenzie et al., 2011), and metformin treatment in diabetic patients significantly reduced the risk of CRC compared with the control group (Tseng, 2012). Metformin usage in CRC patients with diabetic mellitus showed a 40% improvement in overall survival when compared with patients treated with the other anti-diabetic agents (Garrett et al., 2012). In accordance with these studies of reduced cancer risk, metformin is activating AMP-activated protein kinase (AMPK) which is a central master of cellular energy metabolism, cell survival and proliferation (Griss et al., 2015). In preclinical studies, metformin exerts anti-proliferative effect in diverse cancer cells including CRC cells and inhibits tumor growth in vivo xenograft studies (Buzzai et al., 2007; Nangia-Makker et al., 2014). Moreover, many researchers showed that combined treatment of metformin with other chemotherapeutic and targeted agents synergistically increases the anticancer effect, suggesting the possibility to reduce the dose of drugs with severe toxicity (Iliopoulos et al., 2011; Zhang & Guo, 2016).
In our previous studies using HCT15 CRC cells with coexistent mutations of KRAS and PIK3CA, we showed that metformin treatment decreases the level of pERK, one of the key regulatory components in RAS/RAF/MEK/ERK signaling pathway, and dual PI3K/mTOR inhibitor BEZ235 induces the inhibition of cell proliferation (Oh et al., 2016; Lee et al., 2017). Therefore, we hypothesized that the combination of metformin with BEZ235 could not only potentiate the anti-tumor activity as compared to single agent treatment, but also reduce serious side effects resulting from the combinational regimen which co-targets the two signaling pathways by using other inhibitors with severe toxicity. In the present study, we investigated 1) whether the combination of metformin with BEZ235 could lead to dual pathway inhibition, 2) whether the combination treatment regime would be more effective than either agent alone in inhibiting cell proliferation, and 3) whether the combination would change the pattern of cell cycle distribution in HCT15 CRC cells.
MATERIALS AND METHODS
The human colorectal cancer cell line HCT15 was purchased from American Type Culture Collection (Rockville, MD, USA). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% (vol/vol) heat inactivated fetal bovine serum (Gibco BRL, Rockville, MD, USA) and 1% streptomycin/penicillin at 37℃ in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were maintained mycoplasma free by treating 5 µg/mL of Plasmocin (InvivoGen). BEZ235 was obtained from LC laboratories (Woburn, MA). The compound was initially dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich, St. Louis, MO) to a concentration of 1 mM and further diluted in DMEM media. Metformin (also known as 1,1-dimethylbiguanide hydrochloride) was purchased from Sigma-Aldrich and dissolved in DMEM media to a working concentration of 100 mM.
MTT assay was applied to measure cell viability as described previously (Lee et al., 2017). Briefly, cells were harvested and seeded in 24-well plates at a concentration of 5×104 cells/well for 24 hr. Then, cells were treated with increasing concentrations of BEZ235 (12.5-100 nM), metformin (0.25-2 mM), their combinations or vehicle control for 48 hr. Experiments were performed in triplicate, each conducted in quadruplicate. The IC50 values (concentrations of drugs resulting in 50% decrease in cell viability relative to controls), combination index (CI) and drug reduction index (DRI) were calculated using CompuSyn software (ComboSyn Inc, Paramus, NJ, USA). The CI value is a quantitative measure of the degree of drugs interaction. According to the recommendation of Chou-Talalay (Chou & Talalay, 1981), CI<1 indicates synergistic effects of drugs; CI=1 indicates additive effect; CI>1 indicates antagonism. DRI denotes how many folds of dose reduction are allowed for each drug due to synergism as compared to the dose of each drug alone.
Western blotting assays were carried out as previously described (Oh et al., 2016). Primary antibodies included pERK1/2 (Tyr204), ERK1/2, cyclin D1, cyclin B1 (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), and pRb (Ser807/811), p27 Kip1, p4E-BP1 (Ser65), 4E-BP1, pS6 (Ser240/244), S6 (all from Cell Signaling Technology, MA, USA). Following incubation with secondary antibodies conjugated to horseradish peroxidase (Cell Signaling), immunoreactivity was detected with enhanced chemiluminescence method (Santa Cruz Biotechnology).
Flow cytometry to determine cell cycle distribution was performed as previously described (Lee et al., 2017). Briefly, cells were plated in six-well plates and treated with different concentrations of BEZ235, metformin, their combination and vehicle control for 48 hr. Cells were harvested and fixed overnight in 50% ethanol at 4℃. Fixed cells were washed with cold PBS and incubated with RNase (200 µg/mL) for 30 min at 37℃, and followed by propidium iodide staining. Cell cycle distribution was assayed using BD FACSCaalibur Flow Cytometry System and data were analyzed with CellQuest software (Becton Dickinson).
Cells were plated in 6-well culture dishes at a density of 200 cells per well. After 24 hr, cells were treated with BEZ235, metformin and their combination. Every three days, medium was changed with fresh medium containing the corresponding concentration of the drugs. Following twelve-day treatment, cell colonies were washed with cold PBS and then fixed with ice-cold 100% methanol and pictures were taken with a digital camera (Olympus).
RESULTS
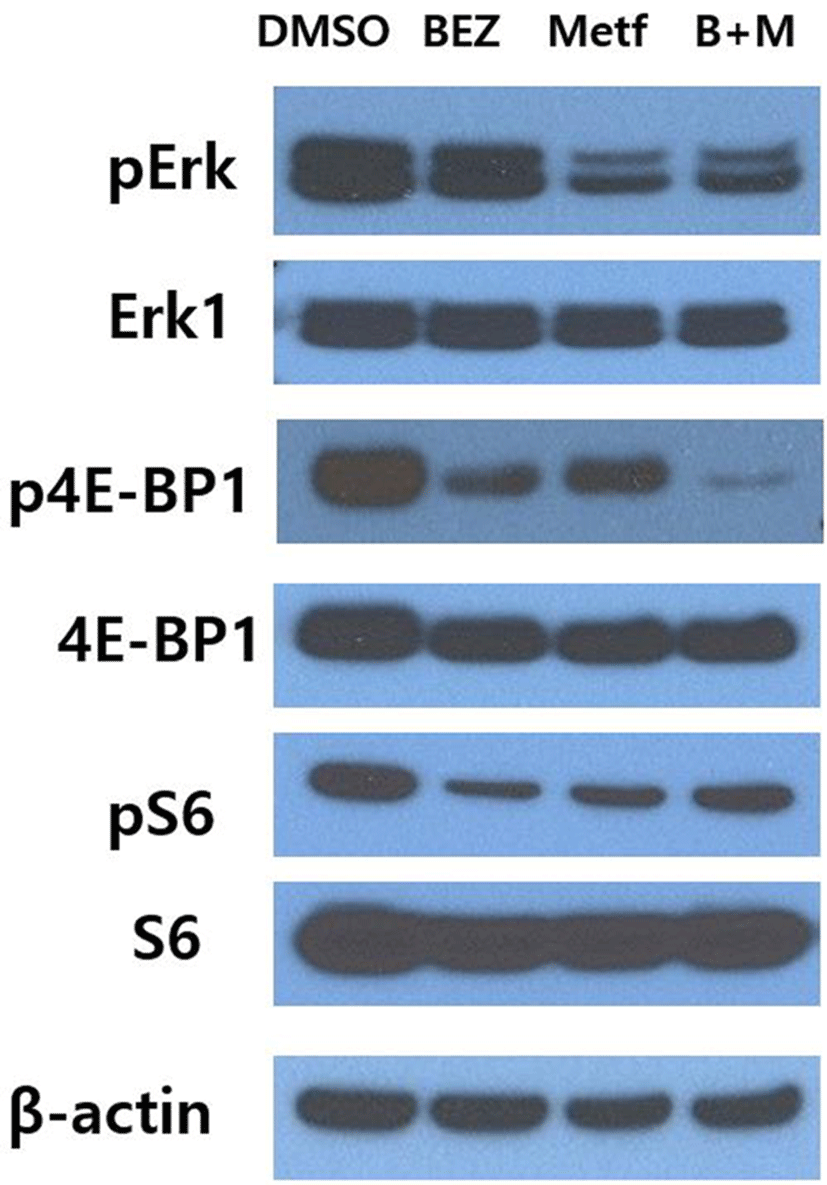
To investigate whether metformin could affect the response of HCT15 CRC cells to BEZ235, we first monitored the alterations of the main downstream effector proteins of PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways following treatment of metformin, BEZ235 or their combination for 24 hr. Single agent metformin showed significant effects on both pathways, where it markedly inhibited the phosphorylation of ERK of RAS/RAF/MEK/ ERK pathway as well as the phosphorylation of S6 and 4E-BP1 of PI3K/AKT/mTOR pathway (Fig. 1). On the other hand, single agent BEZ235 exerted little effect on the level of pERK, whereas it led to a significant inhibition of phosphorylation of S6 and 4E-BP1. The combination of BEZ235 and metformin showed a synergistic suppression of 4E-BP1 phosphorylation. However, the combination of BEZ235 and metformin did not lead to such a synergism in the levels of pERK and pS6 (Fig. 1).
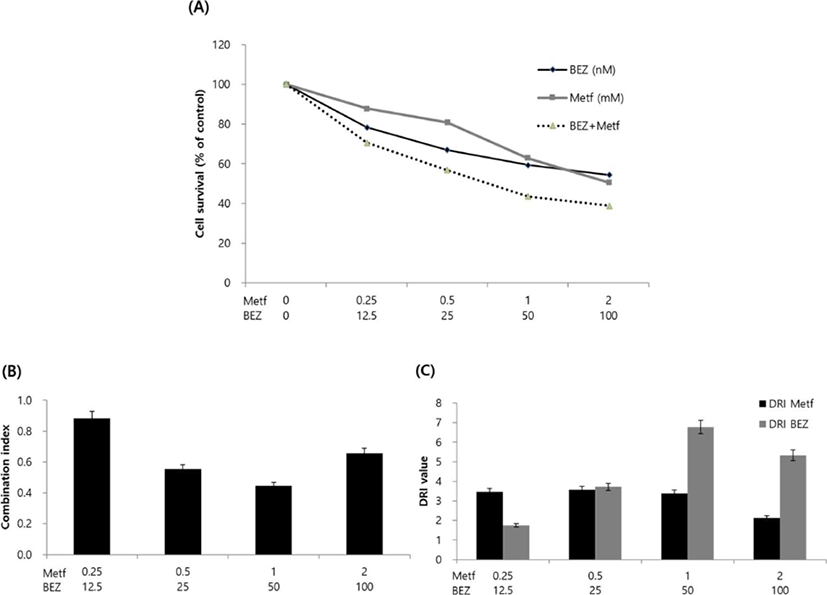
Next, we analyzed the effect of metformin, BEZ235 or their combination on cell viability. HCT15 cells were incubated with various concentrations of drug alone or with their combinations for 72 hr, and cell viability was determined using MTT assay. Treatment with metformin or BEZ235 resulted in the inhibition of cell viability in a concentration-dependent manner. IC50 values (concentrations of drugs leading to 50% decrease in cell viability relative to controls) for metformin and BEZ235 were 2.47 mM and 180.25 nM, respectively. The combined treatment of the two drugs showed more decrease in cell viability as compared to those obtained from treatment of metformin or BEZ235 alone (Fig. 2A).
To characterize the response of HCT15 CRC cells to the combination of metformin and BEZ235, we combined two drugs in a constant ratio to each other and calculated the CI and DRI using CompuSyn software. CI values ranged from 0.44 (for the combination of 1 mM metformin and 50 nM BEZ235) to 0.88 (for the combination of 0.25 mM metformin and 12.5 nM BEZ235) and the CI value at ED50 was 0.68, which indicate synergism according to the method of Chou-Talalay (Chou & Talalay, 1981, Fig. 2B). We also found that the DRI values were always above 1 at any combination of two drugs (Fig. 2C).
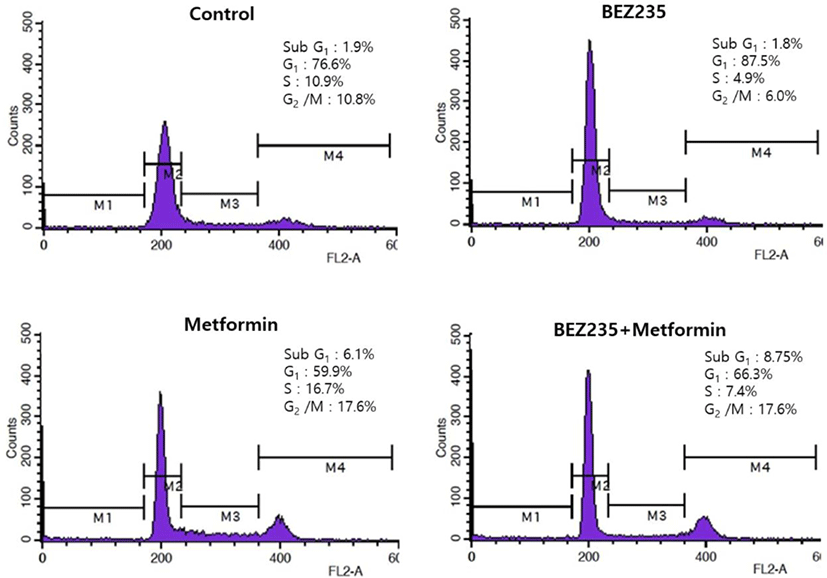
To delineate whether BEZ235, metformin and their combination exert growth inhibitory effects via the changes of cell cycle progression, we analyzed the cell cycle distribution by flow cytometry using PI staining, compared with the vehicle-treated control cells. HCT15 CRC cells treated with BEZ235 for 48 hr displayed a prominent cell cycle arrest in G1 phase with decrease of the cell population in S and G2/M phases. On the other hand, metformin caused a reduction of the cell population in G1 phase from 76.6% of control to 59.9%. The decrease of cell population in G1 phase is accompanied by an increase of the cell population in S and G2/M phases, suggesting that metformin induces cell cycle arrest in S and G2/M phases. Of note, metformin led to a significant increase of sub-G1 cell population from 1.9% of control to 6.1%, raising a possibility of apoptosis induction. Interestingly, the pattern of cell cycle distribution following BEZ235 and metformin co-treatment showed a remarkable reduction of cell population in S phase and increase of sub-G1 cell population as compared with metformin-treated cells (Fig. 3).
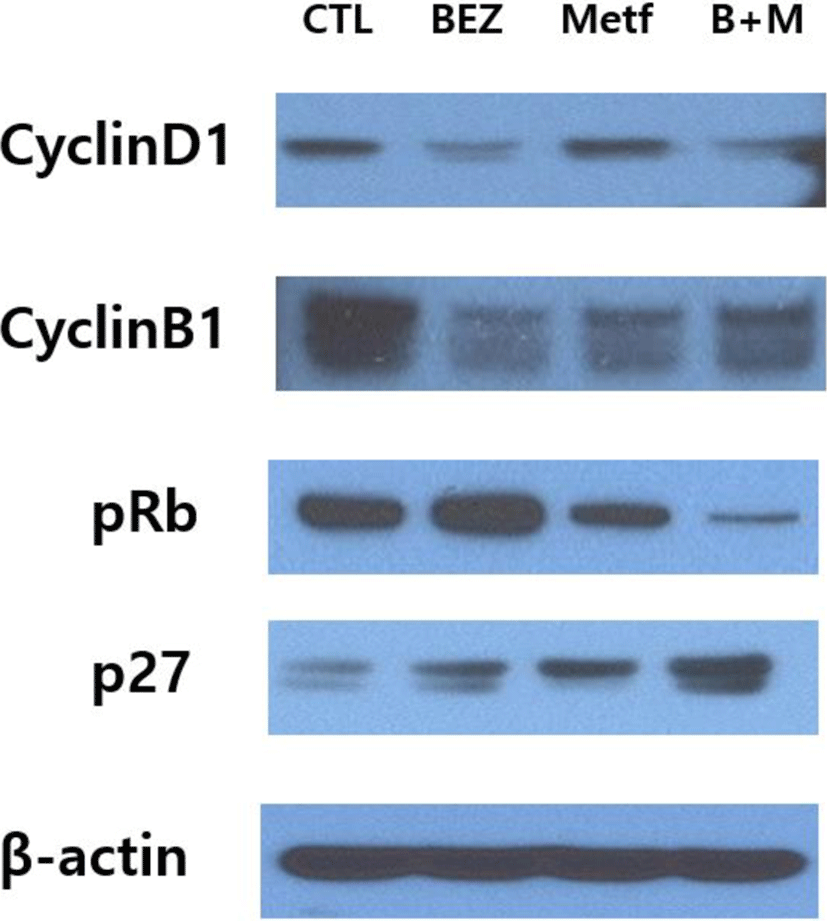
We then examined the regulation of proteins involved in cell cycle by using immunoblot assay. Expression of cyclin D1 was down-regulated in HCT15 cells treated with BEZ235 or the combination of BEZ235 and metformin, but treatment with metformin alone did not show any significant change. The remarkable decrease in cyclin B1 levels was observed in cells treated with BEZ235, metformin and their combination. The phosphorylation of retinoblastoma tumor suppressor protein (pRb) was increased by BEZ235 and reduced by metformin, while their combination markedly suppressed the level of pRb. P27Kip1, a member of the Cip/Kip family of cyclin-dependent kinase (cdk) inhibitors, was upregulated by BEZ235 and metformin, and the effect was further enforced by the combination of BEZ235 and metformin (Fig. 4).
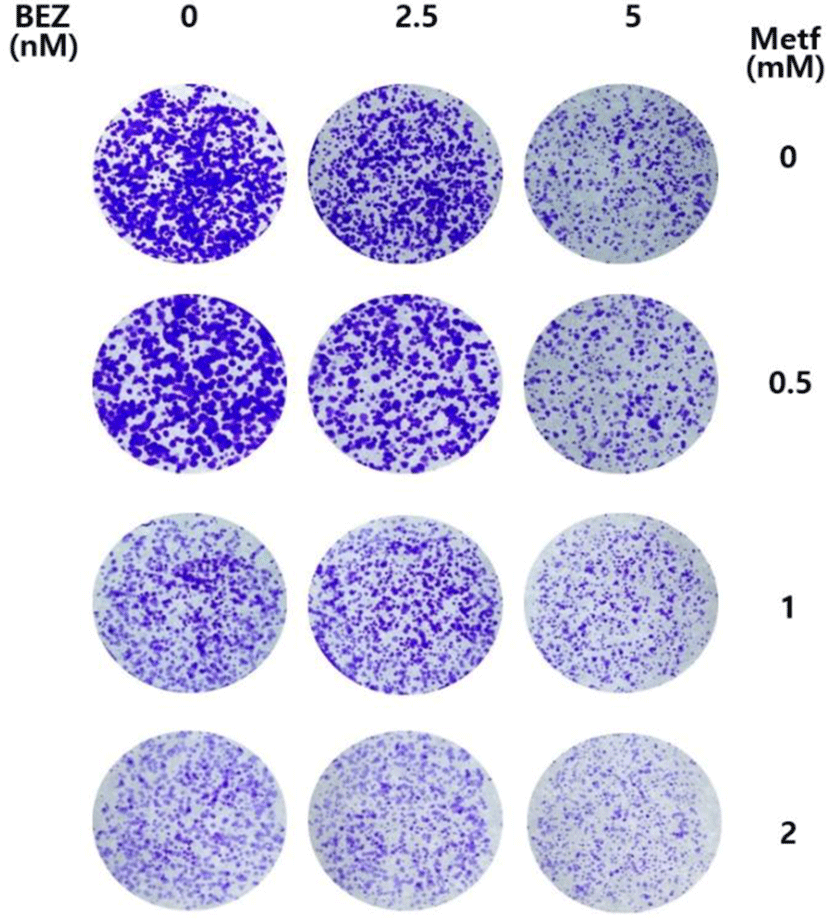
We investigated whether metformin augments the antiproliferative activity of BEZ235 in the long term culture system using colony formation assay. As shown in Fig. 5, BEZ235 or metformin alone at the tested doses in a 12-day culture resulted in a partial inhibition of colony formation. On the other hand, the combined treatment with BEZ235 and metformin potentiated the inhibitory effect on the formation and growth of colonies when compared with either agent alone. Thus, these results further support the synergistic anti-proliferative effect of BEZ235 and metformin in cell survival assay.
DISCUSSION
Cell survival and proliferation are mainly dependent on the regulation of RAS/RAF/MEK/ERK and PI3K/AKT/ mTOR cell signaling pathways, and alteration of downstream components of these signaling cascades, either through somatic mutation or epigenetic modification is often implicated in tumorigenesis (Liu et al., 2009; Santarpia et al., 2012). Although drugs targeting these pathways, developed and approved for treatment of cancer patients, showed remarkable responses in certain cancer types, many challenging issues such as drug resistance and side effects remain unresolved. Mechanistic studdies revealed the drug resistance is related to a negative crosstalk between two signaling pathways (Mendoza et al., 2011). Several preclinical studies using various cancer cells demonstrated that combined targeting of these two pathways is more effective in suppression of cancer cell proliferation compared to the single treatment groups (Britten, 2013; Posch et al., 2013). However, clinical trials of this co-targeting strategy achieved tumor regression to varied extent between 25 and 64%, and the combination therapy caused severe drug-related toxicity in colorectal cancer patients (Shimizu et al., 2012).
In this study, we showed that metformin combined with BEZ235 is able to synergistically increase the anti-tumor activity in HCT15 CRC cells harboring both KRAS and PIK3CA mutations. CRC patients with KRAS mutations showed poorer overall survival and increased risk of relapse, especially when the conventional first-line chemotherapies have failed (Andreyeb et al., 1998). Moreover, targeted therapies against the epidermal growth factor receptor (EGFR) using cetuximab and panitumumab provide no benefit to CRC patients with KRAS mutations, although anti-EGFR treatment improves the response rate and overall survival in patients with KRAS-wild type CRC (Lievre et al., 2006; Benvenuti et al., 2007; Amado et al., 2008; Gong et al., 2016). Consequently, great effort has been made to develop drugs directly targeting KRAS and indirectly inhibiting the downstream effectors such as BRAF and MEK. To date, however, drug development to directly inhibit mutant RAS has failed. Monotherapy or combination therapy with inhibitors of BRAF and MEK did not show clinical efficacy in patients with KRAS mutation (Bennouna et al., 2011; Gong et al., 2016). Our previous studies using HCT15 CRC cell showed that metformin suppresses the phosphorylation of ERK and induces cell cycle arrest in S phase (Lee et al., 2017), and BEZ235, a dual inhibitor of PI3k and mTOR, produces G1 cell cycle arrest (Oh et al., 2016). Therefore, we reasoned that a combination of metformin and BEZ235 could provide more effective strategy to inhibit cell survival and proliferation in HCT15 CRC cell, and showed that the combination results in a synergistic inhibition of cell growth. We confirmed that the synergy is unlikely through enhanced suppression of pERK activation. Instead, our experimental results suggest that the combination induces synergistic inhibition of 4E-BP1 phosphorylation, implying the underlying mechanism of synergy may be related to the enhanced suppression of PI3K/AKT/mTOR pathway rather than that of RAS/RAF/MEK/ERK pathway. In addition, our cell cycle analysis with flow cytometry showed that treatment with BEZ235 and metformin leads to G1 and G2/M arrest, respectively. Thus, another possible explanation for the synergistic effect would be that HCT15 cells with primary resistance to BEZ235 and thus escaping from G1 arrest are captured in the G2/M phase in response to metformin.
Metformin is a widely prescribed and well-tolerated drug for type II diabetes melitus. In cancer cells, metformin disrupts mitochondrial complex I (NADH dehydrogenase), leading to an increase in intracellular AMP/ATP ratio. The increased AMP/ATP ratio in turn activates AMPK (AMP-activated protein kinase), which results in inhibition of mTOR and subsequent suppression of the phosphorylation of its two downstream effectors, the 70 kDa ribosomal protein S6 kinase (p70S6K) and eukaryotic initiation factor 4E-binding protein (4E-BP1). Therefore, it has been suggested that the anticancer effect of metformin is mediated via interference with PI3K/AKT/mTOR pathway rather than RAS/RAF/MEK/ERK pathway (Mihaylova & Shaw, 2011; Quinn et al., 2013). Here, our present result showed that metformin reduces the phosphorylation of ERK1/2 as well as S6 and 4E-BP1. We considered that metformin might increase the phosphorylation of ERK1/2 since these two pathways has been known to negatively regulate each other's activity via cross-inhibition (Mendoza et al., 2011). At present, we have no idea on this contradictory outcome. However, there are several studies showing the reduction of pERK in response to metformin (Niehr et al., 2011; Mohammed et al., 2013). Metformin leads to a dramatic reduction in epidermal growth factor activation and other receptor tyrosine kinases such as human epidermal growth receptor 2, which can inhibit the signaling through downstream pathways including the phosphorylation of ERK (Memmott et al., 2010; Ma et al., 2014). Metformin also suppressed tumor cell migration and invasion through inhibiting the phodphorylation of ERK (Hsieh et al., 2014). Of note, Niehr et al. (2011) and our unpublished in vitro studies showed that synergy obtained from combination of metformin with other targeted drugs is associated with reduction of pERK following metformin treatment. Thus, we tentatively suggest that the suppression of pERK in response to metformin could serve as a biomarker to predict potential synergism in the anticancer therapeutics using drug combination with metformin.
Taken together, our study provides evidence that the combination of an inhibitor of PI3K/AKT/mTOR pathway with metformin synergistically induces anticancer effects in HCT15 colorectal cancer cells with mutations of both KRAS and PIK3CA via suppression of pERK. Additional studies are needed to delineate the in vivo effect of our combination strategy, but our study suggests a possible therapeutic option for treatment of colorectal cancer, especially harboring KRAS mutation.