INTRODUCTION
ES cells are pluripotent cells derived from the inner cell mass (ICM) of embryos (Evans & Kaufman, 1981; Thomson et al., 1998). ES cells are able to self-renew in an unlimited and symmetrical manner and have the capability of differentiation to multiple cell lineages in vitro (Brook & Gardner, 1997; Nagy et al., 1990). Potentials of ES cells have positioned ES cells as a good model system, therefore now ES cells are widely used for studying molecular mechanisms involved in self renewal/differentiation and development, cell therapy, and drug screening (Bain et al., 1995; Lerou and Daley, 2005; Sartipy et al., 2007). To facilitate these studies, a rapid and effective gene transfer method is needed. Several techniques have been adopted to deliver genes into ES cells until now; electroporation (Mamo et al., 2010), liposome-based transfection methods (Ko et al., 2009), nucleofection (Lakshmipathy et al., 2004), viral transfection (Gropp et al., 2003; Ma et al., 2003), and magnetofection (Lee et al., 2008). However, generally the transfection efficiency is not high. Furthermore, there is a pitfall also in expression of foreign genes in ES cells. Major constraint is that integration into the genome is poor and the exogenous gene is often silenced even when it has been successfully integrated into the genome. For example, in the case of transfection with retroviral vector, DNA methylation in the LTR leads to retrovirus silencing and defines the promoter region CpGs as a repressive element in ES cells (Swindle et al., 2004). In addition, ES cells tend to differentiate during the selection procedure and obtaining a reasonably pure cell line is very difficult (Wiles & Johansson, 1999).
To regulate expression of a specific gene, cells have to finely control the coiling and uncoiling of DNA around histones. Acetylation and deacetylation of histones contribute to the epigenetic regulation (Grunstein, 1997). There are two classes of enzymes involved in determining the state of histone acetylation, histone acetyl transferases (HAT) and histone deacetylse (HDAC). HDAC inhibitors induced changes in the acetylation status of chromatin and other non-histone proteins, leading to changes in gene expression (Marks et al., 2000).
Trials to improve the efficiency of gene transfer and gene expression using HDAC inhibitors have been performed in various cells. It was reported that HDAC inhibitors enhance the transcription of adenoviral transgenes in cancer cells (Dion et al., 1997; Goldsmith et al., 2003; Kitazono et al., 2001). For example, a HDAC inhibitor FK228 has the capability to augment adenoviral transgene expression in several different cancer cell lines (Goldsmith et al., 2003). Adenoviral transgene products were amplified by sodium butyrate (NaB: 0.5-5 mM) and trichostatin A (TSA: 0.1-1 μM) in HeLa and A549 cells (Dion et al., 1997). According to a recent study, HDAC inhibitors such as TSA, valproic acid (VPA) and OSU-HDAC42 enhance the expression of genes under the control of a CMV promoter in vitro and in vivo (Lai et al., 2010). Considering that the combined treatment of HDAC inhibitors with 5-Aza-dC (inhibitor of DNA methylase) induces synergistic activation of a transgene, it is likely that there is a cross-talk between histone acetylation and DNA methylation (Choi et al., 2005).
Here, we tested the effect of HDAC inhibitors on transfection in mouse ES cells and found that HDAC inhibitors enhance the transgene expression. In addition, we further enhanced gene delivery and transgene expression by modifying transfection condition.
MATERIALS AND METHODS
R1 mouse ES cells were maintained on irradiated mouse embryonic fibroblast (MEF) cells in ES medium which contains DMEM (Hyclone, Logan, UT), 15% fetal bovine serum (Hyclone), 2 mM L-glutamine, 0.1 mM β-mercaptoethanol (Sigma-Aldrich, St. Louis, MO), and 1% nonessential amino acids (Gibco, Carlasbad, CA). Mouse ES cells were detached from the plates by incubation in TrypLE (Gibco) solution for 3 min at 37°C in a 5% CO2 atmosphere and then harvested via centrifugation. Mouse ES cells were washed with medium and then moved into the MEF plated dish and maintained at 37°C in a 5% CO2 incubator.
R1 cells were cultured on 0.1% gelatin-coated plates in ES medium supplemented with leukemia inhibitory factor (LIF, 10 ng/ml). R1 cells were harvested using TrypLE solution and then 4×105 cells were seeded into 0.1% gelatin-coated 12 well plates 6 hr, 9 hr, 12 hr, and 16 hr prior to transfection. The pMSCV-neo-CMV-GFP (pMSCV-GFP) construct was generated in this study using pMSCV-neo as a parental vector (Grez et al., 1990; Miller and Rosman, 1989). The pMSCV-GFP vector was isolated using Qiagen plasmid MIDI-prep kit (Qiagen, West Sussex, UK).
For transfection using the Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA), 1.6 μg of plasmid DNA (pMSCV-GFP) and 4 μl of Lipofectamine™ 2000 reagent were mixed with 100 μl each of opti-MEM for 5 min at room temperature. Two solutions were then mixed and incubated for 20 min. This complex was added into antibiotics-free ES medium in each well of 12 well plates. For transfection using the FuGENE® HD (Roche, Indianapolis, IN, USA) reagent, 1.0 μg DNA and 3 μl of FuGENE® HD were applied to each well of 12 well plates and the plates were incubated for 15 min at room temperature. After transfection with Lipofectamine™ 2000 or FuGENE® HD, HDAC inhibitors were added at the indicated concentration and the plates were incubated for 1 day at 37°C in a CO2 incubator before analysis for the GFP or stem cell marker expression. HDAC inhibitors (Sigma-Aldrich) used in this study are as followings: trichostatin A (TSA), sodium butyrate (NaB), and valproic acid (VPA).
The expression of GFP was assessed by measuring fluorescence using FACScan. For staining of stem cell markers, control and trasnfected ES cells were fixed and permeabilized by cytofix/cytoperm™ (BD Biosciences, Bedford, MA). The cytofix/cytoperm™ wash solution was added to the cells together with individual primary antibodies to Oct-3/4 (BD Biosciences), Sox-2, Klf-4, and Nanog (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and incubated for 1 hr. After washing with cytofix/cytoperm™ wash solution, the cells were incubated with the FITC-conjugated or Alexa 594-labeled secondary antibody (Invitrogen, Carlsbad, CA, USA) in cytofix/cytoperm™ wash solution at 4°C in the dark for 1 hr. After washing, samples were analyzed with FACScan.
Total RNA was isolated using TRI reagent®, according to the manufacturer’s protocol (MRC, Cincinnati, OH, USA). The final pellet was dissolved in 20 μl of diethyl-pyrocarbonate (DEPC)-treated distilled water. 5 μg of total RNA was reverse-transcribed in the first-strand buffer containing 6 μg/ml oligo (dT) primer, 50 U M-MLV reverse transcriptase (Invitrogen), 2 mM dNTP, and 40 U RNaseOUT™ recombinant ribonuclease inhibitor (Invitrogen). The reaction was conducted at 42°C for 1 hr. One microliter of the cDNA synthesis was subjected to the standard PCR reaction for 20-30 cycles of denaturation for 60 sec at 95°C, annealing for 60 sec at 58°C, and elongation for 60 sec at 72°C. The primer sequences were: GAPDH, 5’-ACCA CAGTCCATGCCATCAC-3’ (sense) and 5’-TCCACCA CCCTGTTGCTGTA-3’ (anti-sense) (452 bp); Oct4, 5’-CTCGAACCACATCCTTCTCT-3’ (sense) and 5’- GGC GTTCTCTTTGGAAAGGTGTTG-3’ (antisense) (313 bp); Nanog, 5’- AGG GTC TGC TAC TGA GAT GCT CTG -3’ (sense) and 5’- CAA CCA CTG GTT TTT CTG CCA CCG -3’ (antisense).
The transfected cells were cultured in the presence of 250 μg/ml G418 (Sigma-Aldrich) and G418 resistant cells were selected for 2-3 weeks. Cells were maintained on 0.1% gelatin-coated dish in ES culture medium supplemented with LIF. After culture for 3 weeks, GFP expression was determined by measuring fluorescence using FACScan (BD Biosciences). Expression of stem cell markers such as Oct-3/4, Sox-2, and Klf-4 was assessed by flow cytometry using FACScan.
RESULTS AND DISCUSSION
Several previous reports suggest that the efficiency of gene transfer and gene expression can be enhanced by HDAC inhibitors. Therefore, we investigated the effect of HDAC inhibitors on transfection efficiency in R1 mouse ES cells.
Many cellular and viral promoters, including cellular polypeptide chain elongation factor-1 (EF1), cytomegalovirus (CMV), and the Rous sarcoma virus (RSV) promoters, have been used to drive gene expression in ES cells (Ward and Stern, 2002; Zeng et al., 2003). Individual promoters have different capabilities to direct expression of reporter gene expression in ES cells and the CMV and EF1 promoters are known to be most effective (Zeng et al., 2003). Here, we constructed an expression vector, pMSCV-GFP, harboring enhanced GFP gene in the downstream of CMV promoter.
HDAC inhibitors such as NaB, VPA, and TSA were treated for 24 hr after transfection using two different transfection reagents, Lipofectamine™ 2000 and FuGENE® HD, and the GFP expression levels were examined by FACS analysis. Population of GFP positive cells increased up to about 180% in the presence of the HDAC inhibitors compared to untreated control (Fig. 1). We obtained similar results when we used either Lipofectamine™ 2000 (Fig. 1A) or FuGENE® HD (Fig. 1B). However, there was some difference in the efficacy of the transfection.
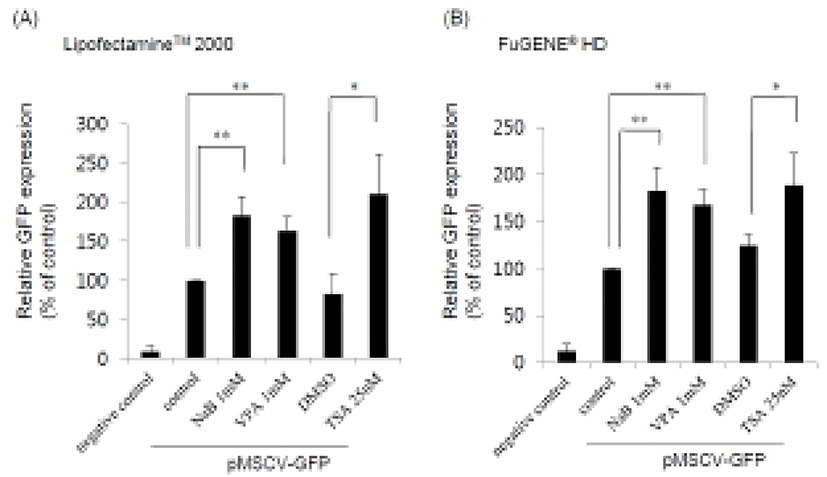
When we checked the percentage of GFP positive cells in the case of Lipofectamine™ 2000, the transfection efficiency in the presence of HDAC inhibitor was observed about 15%. In contrast, a lower percentage of GFP positive cells were obtained by FuGENE® HD treatment (~4%). These data reveal that HDAC inhibitors may be generally used as an enhancer for efficient gene delivery and expression in mouse ES cell. Additionally, we tested another vector including CMV promoter, pEGFP-C2 (Clontech) and found that the transgene expression was also enhanced by HDAC inhibitors (data not shown). This result suggests that enhancement of transgene expression in ES cells can be a general effect of HDAC inhibitors irrespective of the vector types.
According to recent reports in mouse ES cells, DNA methyltransferase and HDAC inhibitors, especially VPA, improve the efficiency of reprogramming mouse embryonic fibroblasts (MEF) using genetic factors (Huangfu et al., 2008a; Huangfu et al., 2008b). TSA treatment after somatic nuclear transfer in mice dramatically improved the efficacy of current cloning technique (Kishigami et al., 2006). VPA overrode Polycomb-mediated silencing of Hoxb gene in mouse ES cells (Boudadi et al., 2013). Also, HDAC inhibitor and DNA methyl transferases alter imprinted gene regulation in ES cells. Histone modification plays an important role in establishing a chromatin state permissive to early embryonic gene expression in ES cells (Baqir and Smith, 2006). In this context, our results showing effect of HDAC inhibitors on transgene expression suggest that HDAC mediated chromatin remodeling may contribute to a low ectopic gene expression in ES cells. As we examined GFP expression after transfection, the outcome can be affected by gene delivery efficiency as well as the extent of the delivered trangene expression. Therefore, it is also possible that modulation of microtubule acetylation through HDAC6 inhibitor leads to increase of gene delivery efficiency as previously reported in human adenocarcinoma A549 cells (Vaughan et al., 2008). For better understanding on this issue, further detailed investigation is required in the future.
Although the transgene expression was increased by the HDAC inhibitors (Fig. 1), the percentage of transfected GFP positive cells is still low. Therefore, to get a better efficiency, we tried optimization of the protocol. As ES cells are inclined to form clumps, exposed surface region of ES cells is limited. We usually transfer ES cells onto a culture plate and culture for 16 hr prior to transfection. Therefore, we tried to adjust the plating time of ES cells: three different plating time periods (6 hr, 9 hr, and 12 hr). HDAC inhibitors were treated at the indicated concentration after transfection by Lipofectamine™ 2000 and FuGENE® HD. As shown in Fig. 2, population of GFP positive cells after transfection using Lipofectamine™ 2000 in mouse ES cells increased by HDAC inhibitors in a dose-dependent manner. Lipofectamine™ 2000 (Fig. 2) and FuGENE® HD (data not shown) produced similar pattern of transfection efficiency even though overall transfection efficiency was higher with Lipofectamine™ 2000. The highest transfection efficiency was appeared when we performed transfection after plating the cells for 6 hr and incubate the cells in the presence of HDAC inhibitors for 24 hr. Thus, this condition was defined as a standard condition for the following experiments. As both of the GFP positive cell population and the mean fluorescence index (MFI) increased simultaneously, it is likely that the modified protocol enhances gene delivery as well as the transgene expression.
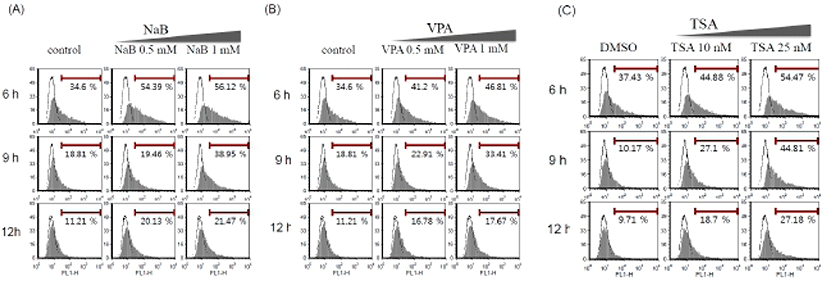
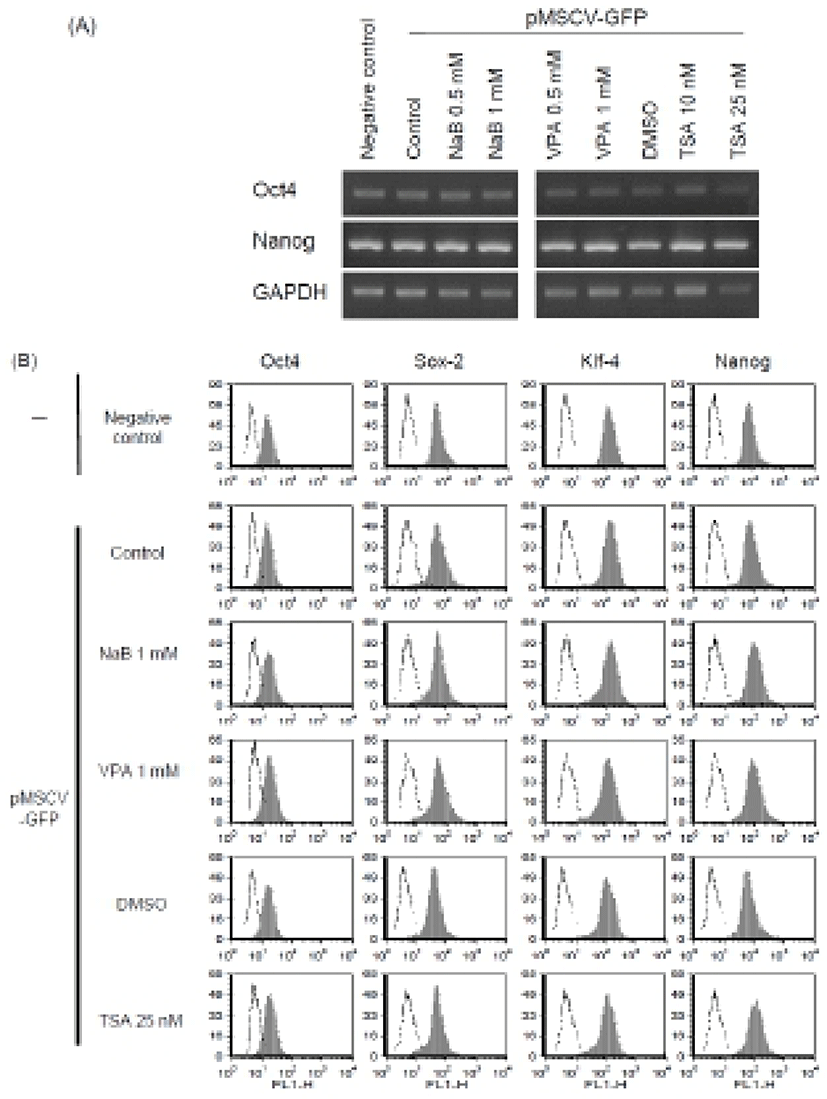
Previous studies have shown that HDAC inhibitors, including TSA, induced the early differentiation and altered characterization of ES cell (Karantzali et al., 2008; Park et al., 2011). It was also reported that mSin3A-HDAC complex is involved in the maintenance of ES cell pluripotency (Baltus et al., 2009). Therefore, to evaluate whether these HDAC inhibitors change the stem cell properties during the transfection procedure in this study, we treated NaB 1 mM, VPA 1 mM, and TSA 25 nM for 24 hr after transfection with Lipofectamine™ 2000 and examined expression of the stem cell markers.
We first confirmed similar expression of stem cell markers Oct4 and Nanog mRNAs in the transfected ES cells (Fig. 3A). Next, we analyzed the expression of stem cell markers such as Oct4, Nanog, Sox-2, and KLF4 after transfection procedure at the protein level. Expression of stem cell markers in the transfected cells was similar with that in the untreated control mouse ES cells as determined by FACS analysis (Fig. 3B). These results suggest that transfection with Lipofectamine™ 2000 and treatment with HDAC inhibitors at the concentration we used permits maintenance of an undifferentiated state of ES cells, at least in the context of stem cell marker expression. It is consistent with previous study reporting that low concentration of NaB (0.1 mM) can be used instead of bFGF: the culture medium including NaB could maintain human ES cells in a feeder free culture condition (Kim et al., 2012; Ware et al., 2009). The human ES cells cultured in the modified condition had normal molecular marker expression compared with control cells. Also, other HDAC inhibitors, such as TSA (10 nM) and VPA (0.5 mM) were shown to support expression of stem cell markers (Kim et al., 2012; Ware et al., 2009). Therefore, it is likely that optimal concentration of HDAC inhibitor doesn’t affect or even enhance the stem cell properties of ES cells.
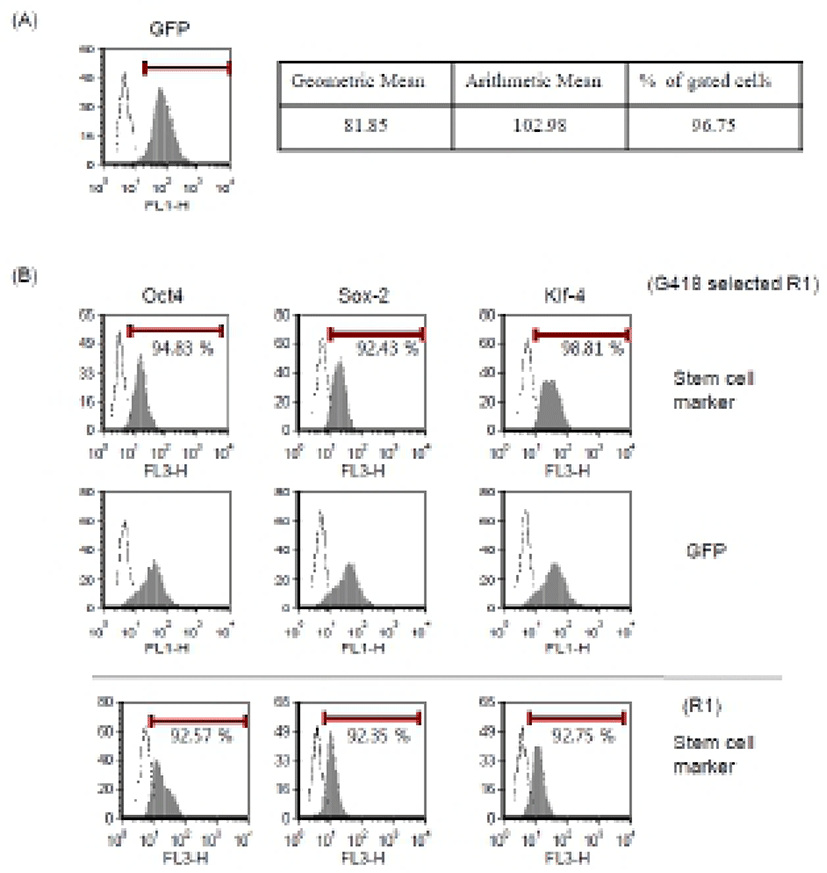
To obtain the stable transgenic ES cells, we next tried to select G418-resistant cells and to verify GFP and stem cell marker expression. Mouse ES cells were transfected with our modified protocol in the presence of NaB. After 24 hr, we changed medium and added 250 μg/ml of G418. G418-resistant cells were selected for 2-3 weeks and expression of GFP was analyzed by FACS (Fig. 4A). These cells were well maintained in vitro and showed typical ES cell characteristics. More than 90% of total cells were positive for the Oct4, Sox-2, and Klf4 markers in FACS analysis, which is comparable to the expression pattern of parental R1 ES cells (Fig. 4B). We also confirmed typical ES cell morphology and positive staining of alkaline phosphatase (data not shown). Therefore, we suggest that the transfection and treatment with HDAC inhibitor do not interrupt stem cell properties of ES cells and the transgenic ES cells maintain their authentic properties.
Taken together, our results suggest that treatment with HDAC inhibitors and modification of plating time can improve the efficiency of transgene delivery and expression in mouse ES cells. Our method will be useful for study and application of ES cells by facilitating genetic modification in mouse ES cells.