
INTRODUCTION
Attempts to target the altered signaling pathways at molecular level have been proven to be effective for the treatment of cancer. Kinases in signal transduction process are closely implicated in tumor cell proliferation and survival, which has led to the development of small kinase inhibitors for cancer intervention. Imatinib, a small-molecule tyrosine kinase inhibitor (TKI) targeting the fusion protein of BCR-ABL kinase found in chronic myeloid leukemia (CML), has been widely used in the CML treatment, exhibiting up to 80% response rate (Druker et al., 1996; Zhang et al., 2009). However, despite this exciting therapeutic efficacy, significant challenges are faced due to drug resistance and side effects of imatinib, resulting in high relapse rate and dose reduction respectively. Although second-generation TKIs such as dasatinib (Hochhaus et al., 2008) and nilotinib (Saglio et al., 2010) have been developed to overcome imatinib resistance and improve the prognosis of CML patients, resistance to these novel inhibitors still arises partly from the emergence of BCR-ABL mutant clones (O’Hare et al., 2011). Therefore, it has been a challenging theme to overcome this compromised effectiveness of imatinib as a single agent for successful treatment outcome in the clinic.
In addition to its inhibition of BCR-ABL tyrosine kinases in CML, imatinib targets selectively other receptor tyrosine kinases such as stem cell factor receptor (c-Kit) and platelet-derived growth factor (PDGF) receptor, and is currently used for the treatment of gastrointestinal stromal tumor (GIST) harboring c-Kit mutation (Joensuu et al., 2001; De Giorgi & Verweij, 2005). Several studies demonstrated that Abl kinases are tyrosine phosphorylated and activated in solid tumors and imatinib promotes apoptosis and inhibits growth of various cancer cells including colorectal cancer (CRC) cells (Attoub et al., 2002; Stahtea et al., 2007; Popow-Wozniak et al., 2011; Abdel-Aziz et al., 2015). Furthermore, recent reports suggested that imatinib suppresses cell proliferation in intestinal adenomas and CRC cell lines via the regulation of Abl-cyclin D1 pathway (Genander et al., 2009; Kundu et al., 2015).
Metformin, a biguanide derivative, is a widely prescribed and well-tolerated first-line drug for type II diabetes mellitus. Metformin lowers the blood glucose level by inhibiting hepatic gluconeogenesis and increasing glucose uptake in skeletal muscles, which lead to a decline in circulating insulin levels (Shaw et al., 2005). Several retrospective studies have described that metformin treatment in diabetic patients significantly reduced the risk of cancer incidence and metformin use in CRC patients with diabetes is associated with decreased mortality, suggesting a potential role of metformin as an anticancer agent (Evans et al., 2005; Lee et al., 2011). In many preclinical studies, metformin suppressed cellular proliferation, caused apoptosis, induced cell cycle arrest, and decreased the incidence and growth of experimental tumors (Isakovic et al., 2007; Ben Sahra et al., 2008; Tomimoto et al., 2008; Alimova et al., 2009). Moreover, combinatory treatment of metformin and other chemotherapeutic drugs showed the synergistic anticancer effects in CRC cells (Zhang et al., 2013; Nanglia-Makker et al., 2014), demonstrating the possibility of decreasing the dose of chemotherapeutic drugs with severe side effects.
The present study was undertaken to investigate whether metformin could be used in conjunction with imatinib to inhibit the growth of HCT15 CRC cells and to delineate its mechanisms of regulation. Herein, we observed synergistic growth inhibition of HCT15 CRC cells with the combination of imatinib and metformin, and described synergistic down-regulation of pERK, cell cycle arrest, and induction of autophagy as the mechanism explaining effects of the combination.
MATERIAL AND METHODS
Imatinib (also known as Gleevec) and metformin were purchased from Sigma-Aldrich (St. Louis, Mo, USA). Imatinib was dissolved in DMSO to a concentration of 50 mM and additionally in DMEM media to a working stock concentration of 100 μM. Metformin was dissolved in phosphate buffered saline to a working concentration of 100 mM. The human colorectal cancer cell line HCT15 was obtained from ATCC (Rockville, MD, USA). The cells were maintained and grown in Dulbecco’s modified Eagle’s medium (1.0 g glucose/L) supplemented with 10% fetal bovine serum (Gibco BRL, Rockville, MD, USA) at 37℃ in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were regularly tested for mycoplasma contamination by treating 5 μg/mL of Plasmocin (Invivo Gen, San Diego, CA, USA). Acridine orange, chloroquine and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetra-zolium bromide (MTT) were all purchased from Sigma-Aldrich.
Cell viability was determined by using MTT method as described previously (Park & Lee, 2014). HCT15 cells were harvested and seeded in 24-well plates at a concentration of 5×104 cells/well, After 24 hr, cells were treated with varying concentration of imatinib (2.5–40 μM), metformin (0.25–4 mM), their combination or vehicle control for 48 hr. Results represent the median of 3 separate experiments each conducted in quadruplicate. The IC50 values, combination index (CI) and dose reduction index (DRI) were calculated by using CompuSyn software (ComboSyn Inc, Paramus, NJ, USA). The resulting CI is a quantitative measure of the degree of drugs interaction. If CI<1, it indicates synergism; if CI=1, it indicates additive effect; if CI>1, it indicates antagonism. DRI denotes how many folds of dose reduction are allowed for each drug due to synergism when compared with the dose of each drug alone.
Western blotting assays were performed as previously described (Song et al., 2011) using the following primary antibodies: cyclin D1, cyclin A, cyclin B1, ERK, pERK (Tyr204) (all from Santa Cruz Biotechnology, Santa Cruz, CA, USA), and LC3B (Cell Signaling, Beverly, MA, USA). After incubation with secondary antibodies conjugated to horseradish peroxidase (Cell Signaling), enhanced chemiluminescence was used for detection of immunoreactivity (Santa Cruz Biotechnology). β-actin (Cell Signaling) served as a loading control.
HCT15 cells were plated onto 60 mm dishes and treated with different concentrations of metformin, imatinib, the combination and vehicle control for 24 hr. Cells were harvested by treating trypsin-EDTA for 5 min at 37℃, washed with PBS and fixed overnight in 50% ethanol at 4℃. Fixed cells were washed with PBS and incubated with RNase (200 μg/mL) for 30 min at 37℃, and followed by propidium iodide staining. Cells were analyzed by using BD FACSCalibur Flow Cytometry System and data were analyzed by using CellQuest software (Becton Dickinson).
To detect acidic vesicular organelles which consist predominantly of autophagosome and autolysosome and appear following autophagy induction, we carried out vital staining with acridine orange after treatment of metformin or imatinib. Briefly cells were washed twice with PBS, stained with acridine orange (1 μg/mL) in HBSS containing 5% FBS for 15 min and then observed with fluorescence microscope.
Cells were seeded in 6 well plates at a density of 200 cells per well. On the second day, cells were treated with metformin or imatinib. Every three days, medium was changed with fresh medium containing the corresponding concentration of the drugs. After twelve day treatment, cell colonies were washed twice with cold PBS and then fixed with ice-cold 100% methanol. Cell colonies were stained with 0.1% crystal violet in 20% methanol and taken pictures with digital camera.
RESULTS
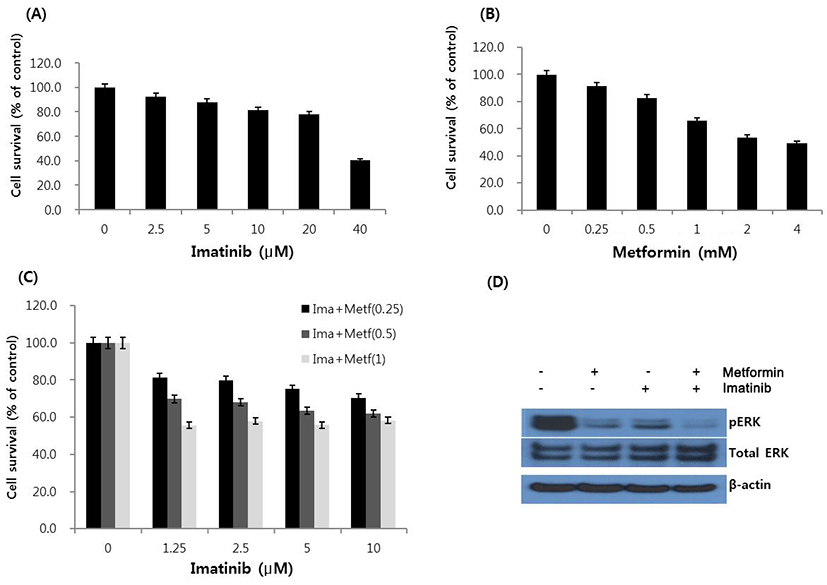
To evaluate the response of HCT15 CRC cells to imatinib and metformin, we first treated HCT15 cells with various concentrations of imatinib and metformin for 48 hr, and cell viability was determined by using MTT assay. As shown in Fig. 1A and B, imatinib and metformin inhibited cell viability in a concentration-dependent manner. IC50 values of imatinib and metformin were 38.2 μM and 2.7 mM, respectively. Next, we analyzed the effect of imatinib in combination with metformin on cell growth by deter-mining the type of interaction between two drugs. Imatinib and metformin were combined in non-constant ratios, in which the fixed dose of metformin (0.25, 0.5 or 1 mM) were co-treated with different concentrations of imatinib (1.25–10 μM). As shown in Fig 1C, anti-proliferative activity of imatinib was augmented in proportion to increasing doses of metformin. The combination index (CI) values ranged from 0.51 to 0.86, indicating synergism according to the method of Chou-Talalay (Chou & Talalay, 1981) (Table 1). The combination also showed that the values of drug reduction index (DRI) were always above 1 at any combination points of two drugs (Table 2). We next screened the change of pERK protein, one of the key regulator proteins in signal transduction pathway, by Western blot assay. Metformin and imatinib each induced a remarkable reduction in pERK, and the combination of metformin and imatinib caused almost complete suppression of pERK level (Fig. 1D).
| Ima (μM) | 1.25 | 2.5 | 5 | 10 | Type of interaction |
|---|---|---|---|---|---|
| CI at 0.25 mM Metf | 0.52 | 0.56 | 0.58 | 0.71 | Synergistic |
| CI at 0.5 mM Metf | 0.51 | 0.52 | 0.52 | 0.66 | Synergistic |
| CI at 1 mM Metf | 0.51 | 0.6 | 0.63 | 0.86 | Synergistic |
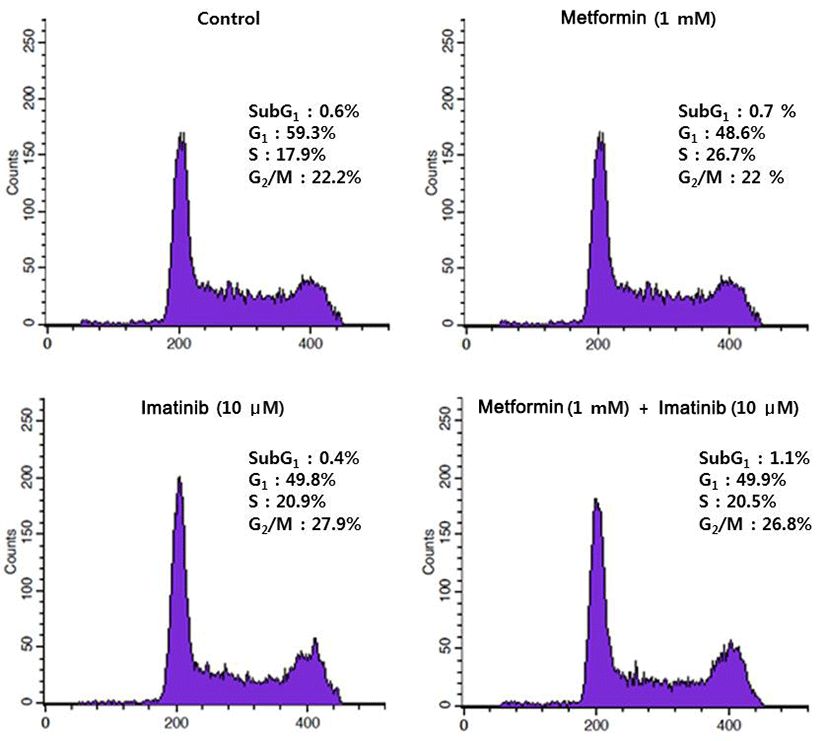
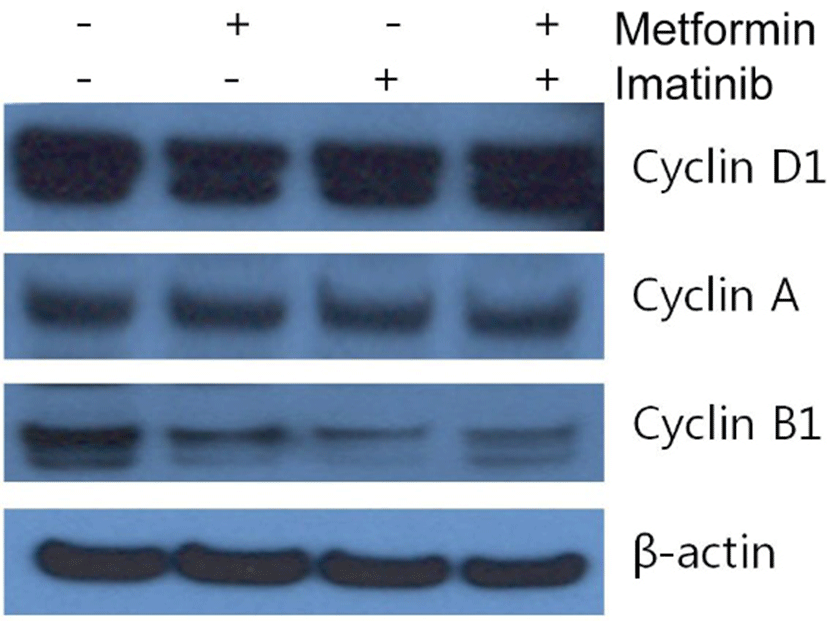
Since imatinib, metform and their combination decreases cell viability, we explored whether these results are attributed to the changes of cell cycle distribution. Cell cycle analysis of HCT15 cells by flow cytometry using PI staining is shown in Fig. 2. After treatment of metformin (1 mM) alone for 24 hr, the percentage of HCT15 cells in S phase was increased from 17.9 % of control to 26.7 %. The increase of cell population in S phase is accompanied by a reduction in G1 phase, indicating that metformin induced cell cycle arrest in S phase. Imatinib (10 μM) caused cell cycle arrest in G2/M phase and to lesser extent in S phase. Combination treatment of metformin and imatinib resulted in similar pattern of cell cycle distribution as treated with imatinib alone. We further monitored the changes of proteins involved in cell cycle regulation using Western blot. As shown in Fig. 3, while there is no apparent alteration in cyclin D1 and A as compared to control, cyclin B1 levels were remarkably reduced by imatinib, metfromin and their combination treatment.
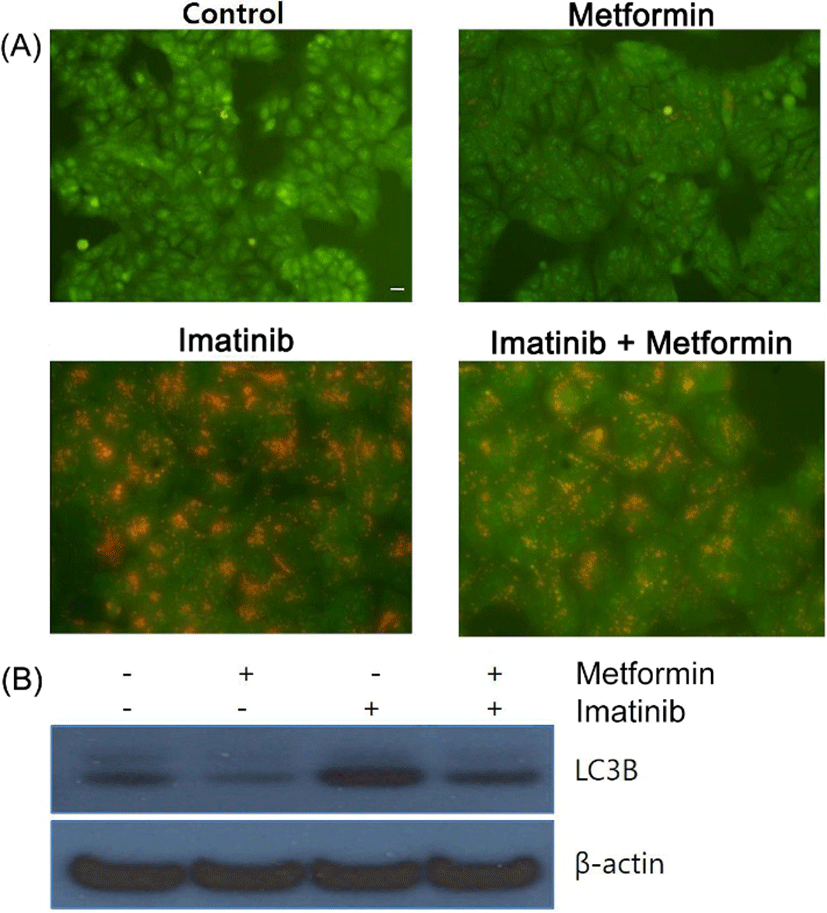
Substantial evidence has been accumulating to reveal that most of chemotherapeutic and molecular targeted agents employed in cancer therapy cause the activation of autophagy in cancer cells (Sui et al., 2013). Therefore, we examined the effect of imatinib, metformin and their combination on autophagy induction. The lysosomotropic agent acridine orange (AO) accumulates in the acidic vesicular organelles (AVOs) such as autophagosomes and autolysosomes, which are formed after the initiation of autophagy. When excited with blue light, AO emits red fluorescence in AVOs and green fluorescence in cytoplasm and nucleus (Takeuchi et al., 2005). It has been also suggested that the microtubule-associated protein light chain 3 (LC3) is a novel marker of autophagy activation. Upon autophagy induction the cytosolic full-length form of LC3-1 (16 kDa) is processed to the cleaved autophagosome-bound form of LC3-II (14 kDa) through lipidation to be associated with autophagic vacuoles (Li et al., 2013). In our study, metformin alone showed a little increased red fluorescent AVOs formation while imatinib and the combination of metformin and imatinib led to the markedly increased red fluorescent AVOs in the cytoplasm compared to control cells (Fig. 4A). Western blot analysis revealed that metformin did not show the increase of the lipidated LC3-II form, whereas imatinib and the combination treatments showed the increased level of LC3-II form, consistent with the results observed in AO staining (Fig. 4B).
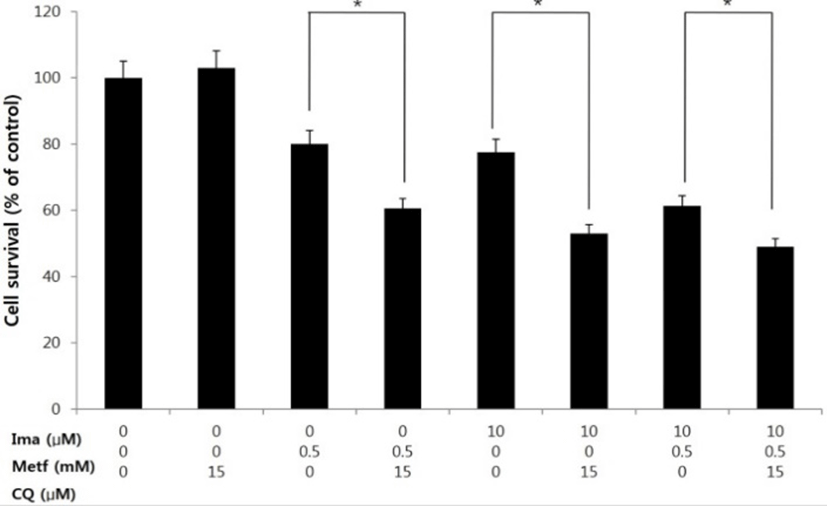
It has been suggested that many anti-cancer therapeutic agents induce autopahgy, which acts as either prosurvival or prodeath role of cancer cells (Yang et al., 2011). Thus we asked how the inhibition of autophagy affects the anti-tumor activity in our treatment regimen. To this end, we employed a lysosmotropic agent chloroquine (CQ), which interferes with the fusion between autophagosomes and lysosome and thus widely used as an autophagy inhibitor (Mizushima et al., 2010). As shown in Fig. 5, co-treatment of metforim, imatinib or their combination with CQ resulted in more decreased cell viability compared to that observed in treatment of metformin, imatinib or combination alone.
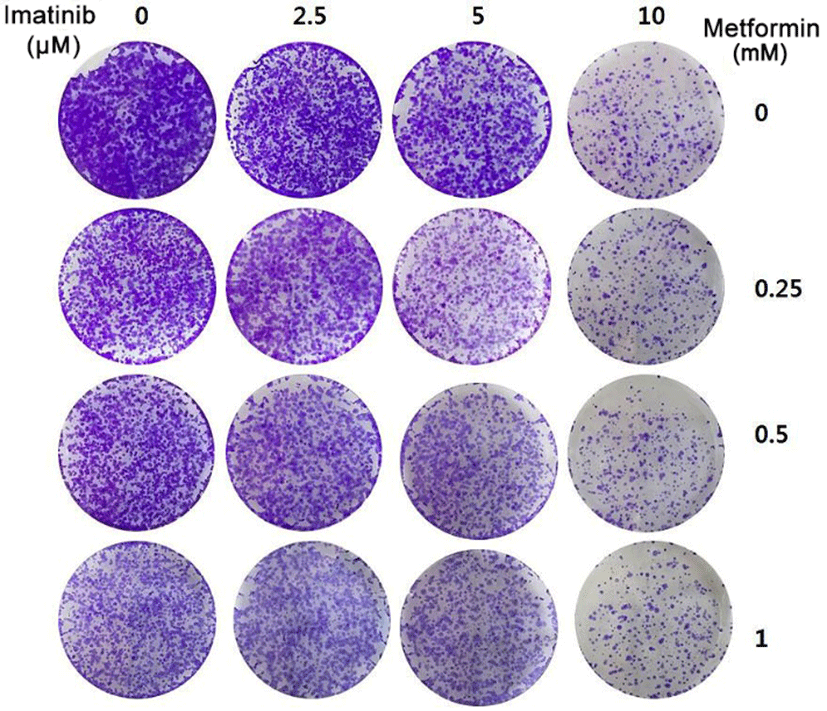
To further confirm the synergistic effect of imatinib in combination with metformin, we tested the long term effect of this drug combination in a 12-day colony formation assay. As shown in Fig. 6, imatinib or metformin alone at the tested doses partially inhibited the growth of colony formation. However the combination of imatinib and metformin potentiated the inhibitory effect on the formation and growth of colonies as compared with either agent alone. Thus, these results further support the synergistic anticancer effect of imatinib and metformin combination treatment in HCT15 CRC cells.
DISCUSSION
The tyrosine kinase inhibitor imatinib has shown a substantial therapeutic activity in patients with chronic myeloid leukemia (CML), a disease resulting from mutated BCR-Abl kinase. Imatinib also inhibits the catalytic activity of PDGF receptor and c-kit receptor kinase. Therefore, imatinib is successfully used for the treatment of GIST with mutated c-kit tyrosine kinase and several studies have suggested that imatinib inhibits cell proliferation and induces apoptosis in CRC cells (Attoub et al., 2002; Stahtea et al., 2007; Popow-Wozniak et al., 2011; Abdel-Aziz et al., 2015). However, despite high response rate of imatinib in CML and GIST patients, significant drug resistance and side effects have become a challenging theme in the clinic. As a candidate to overcome these problems in cancer therapeutics, the biguanide metformin prescribed widely for the diabetes treatment with well tolerable side effects has been tested as a single agent or combination treatment with other drugs since metformin shows the antiproliferative and proapoptotic activity in various cancer cells.
In the present study, we evaluated the effects of metformin, imatinib and their combination in HCT15 CRC cell line, and showed dose- and time-dependent growth inhibitory effect of imatinib and metformin. In addition, the combination resulted in synergistic growth inhibitory effects in HCT15 cells as revealed in CI and DRI values. Recently, Shi et al (2015) reported that metformin also potentiates the anticancer activity of imatinib in CML cells. To elucidate the underlying mechanism for the synergism of the imatinib and metformin combination, we explored changes in the levels of pERK1/2. Interestingly, our study showed that metformin alone significantly suppressed pERK level. Metformin is known to inhibit mitochondrial complex I (NADH dehydrogenase) activity in the electron transport chain, which in turn increases the cellular AMP/ ATP ratio. This high AMP/ATP ratio activates the phosphorylation of AMPK, a master energy sensor within cell, and then pAMPK inhibits mTOR signaling by activating TSC2 and subsequently inhibiting Rheb (Quinn et al., 2013; Wheaton et al., 2014). Therefore we anticipated that metformin might increase pERK, considering that RAS-RAF-MEK-ERK and PI3K-AKT-mTOR pathways negatively regulates each other’s activity and mTOR inhibitorbased therapy often resulted in drug resistance due to the activation of ERK (Mendoza et al., 2011). Currently, the reason is yet unclear, but there are some reports showing the decrease of pE.
RK following metformin treatment (Niehr et al., 2011). In addition, Alimova et al (2009) suggested that the high concentration of metformin inhibits the epidermal growth factor receptor activity, which leads to reduction of pERK in breast cancer cells. The exact mechanism of anticancer activity of metformin at molecular level is a matter of debate, and there is a conflicting issue on the high doses of metformin (1–20 mM) used in vitro cell culture study compared to serum levels (6–30μM) observed in diabetic patients to whom metformin is prescribed. One possible explanation for this may be that cultured cell lines have not the cationic transporter OCT1 to enter metformin into cytoplasm (Niehr et al., 2011). Another is the positively charged metformin is accumulated in tissues or within mitochondria. This might reach the local concentration of metformin up to 1,000 fold higher than the serum levels, similar doses to those used in cell culture models (Wilcock & Bailey, 1994).
Molecular targeted agents for cancer therapeutics are known to induce cell cycle arrest and much attention has been paid to the control of cell cycle in the field of oncology. In our current study, flow cytometry showed that metformin, imatinib and the combination caused an increase in S phase or G2/M phase population 24 hr after treatment. This is in good agreement with the result of western blot analysis showing that cyclin B1 levels were markedly reduced with no apparent changes of cyclin D1 and cyclin A levels after treatments of metformin, imatinib and the combination, since the accumulation of cyclin B1 protein begins at the S phase and reaches the maximal level at G2/M phase (Norbury & Nurse, 1992). In contrast, several studies described that Abl tyrosine kinase regulates cell cycle and blocking of Abl tyrosine kinase with imatinib reduces cell proliferation via depletion of cyclinD1 and hence leads to G1 arrest of cell cycle (Genander et al., 2009). However, a report with CML cells showed that imatinib brings about a transient G1 phase accumulation and S phase depletion peaking at 12 hr after treatment followed by a progressive decrease of G1 phase content and accumulation in S phase at 24 hr (Huguet et al., 2008). Thus, the discrepancy of our results with others seems to reside in the time-dependent changes in cell cycle distribution after imatinib treatment and further detailed analysis is required to delineate the precise temporal pattern of cell cycle distribution after imatinib treatment in HCT15 CRC cells.
Autophagy is a lysosomal catabolic mechanism which is required for the maintenance of cellular homeostasis. Cellular autophagy is induced in environmental or metabolic stress conditions such as nutrient deprivation or serum withdrawal. Many cancer therapeutic agents, either chemotherapeutic or molecular targeted drugs, have been known to induce autophagy and imatinib also activates autophagy in various cancer cells including CRC cells (Abdel-Aziz et al., 2015). Our present study also showed that the anticancer drug imatinib and its combination with metformin activated autophagy in HCT15 CRC cells. Although increasing evidence is accumulating to show the importance of autophagy in cancer therapeutics, the role of autophagy in cancer is still controversial. Autophagy induced following cancer therapy often behaves like a double-edged sword by increasing or diminishing the anticancer activity of drugs. Autophagy induction in response to cancer therapy can act as a cell protective mechanism causing drug resistance of cancer cells. Therefore, inhibition of autophagy can reverse the drug resistance and enhance the cytotoxicity of anticancer drugs. On the other hand, autophagy may give rise to cell death, a second type of programmed cell death (autophagic cell death). In this latter case, autophagy inhibition would increase the viability of cancer cells (Sui et al., 2013). Thus it is important to determine whether autophagy is induced in cancer cells in response to cancer therapy and the induced autophagy serves as cell protective or cell destructive role. In the present study, we showed that inhibition of autophagy induced by metformin, imatinib and the combination augments the cytotoxicity by diminishing the cell viability. This indicates that the induced autophagy acts as cell survival mechanism in HCT15 CRC cells. Our result is consistent with other reports showing that autophagy inhibition by different autophagic inhibitors potentiates the anticancer effect of imatinib in several cancer cells (Crowley et al., 2013).
Taken together, our study reveals that metformin and imatinib inhibits cell viability in dose-dependent manner and metformin synergistically augments the anticancer activity of imatinib in HCT15 CRC cells. Moreover, we showed that autophagy is activated in response to metformin, imatinib and the combination, and the blockage of autophagy resulted in the decrease of cell viability. Thus, our findings suggest that metformin may be used clinically to enhance the antitumor effect of imatinib and inhibition of autophagy can be a potential strategy to increase therapeutic efficacy of imatinib in combination with metformin. Further comprehensive studies are required to examine in vivo synergistic antitumor effect of imatinib and metformin combination therapy for the clinical application.