
INTRODUCTION
Steroidogenesis depends on the availability of cholesterol. For decade, the mechanisms of enzymatic metabolism and transport have been central questions in physiology and developmental biology, as congenital malformations have been linked to defects in steroidogenesis. The first step for steroidogenesis is the transport of cholesterol into the mitochondrial inner membrane cytochrome P450 family 11 subfamily A member 1 (CYP11A1, P450scc; a net turnover number of <20 molecules/one P450scc molecule/second; cytochrome P450 side-chain cleavage enzyme), and then the following steps are mainly at the endoplasmic reticulum (ER). CYP11A1 in mitochondria converts the cholesterol to pregnenolone, and it is metabolized to progesterone by either mitochondrial or ER hydroxy-delta-5-steroid dehydrogenase, 3-beta- and steroid delta isomerase 2 (3 beta-hydroxysteroid dehydrogenase, 3β-HSD, HSD3B2). Steroidogenic enzymes within the ER modify pregnenolone to steroid hormones like progesterone, cortisol, aldosterone, testosterone, and estrogen (Simpson & Waterman, 1988; Papadopoulos & Miller, 2012).
The 30-kDa steroidogenic acute regulatory protein (StAR) was identified with a 37-kDa precursor (STAR-201 based on the alternative splicing form of Ensembl (Dyer et al., 2025) and its gene (Epstein & Orme-Johnson, 1991; Stocco & Sodeman, 1991; Clark et al., 1994; Dyer et al., 2025), as a required protein in the acute steroidogenesis in the testis. It has been accepted that most cholesterol transport depends on the StAR protein (Stocco, 2001; Miller, 2017, 2025). After identifying the STAR gene, STAR expression was also evaluated in nonclassical steroidogenic tissues such as the brain, skin, retina, kidney, etc., along with extending its possible roles (Sugawara et al., 1995; King & Stocco, 2011; Manna et al., 2016). Acute and chronic regulation of steroidogenesis are under the control of trophic hormone stimulation and tissue specificity, and occur in the order of minutes and hours, respectively (Stocco et al., 2005). As expected from the name of StAR, the STAR gene codes for the steroidogenic protein and STAR are required for the rate-limiting step for acute steroidogenesis by delivering cholesterol into mitochondria (Miller, 2025). The chronic response involves the expression of steroidogenic enzymes such as CYP11A1 (Simpson & Waterman, 1988). In a nonsteroidogenic cell, the coexpression of StAR and CYP11A1 system induces steroidogenesis (Lin et al., 1995).
StAR activity is finely modulated through multi-layered mechanisms, including transcriptional control, posttranslational modifications such as phosphorylation, mitochondrial import, proteolytic degradation, and a conformation modification through a partially unfolded molten globule stage (Bose et al., 1999; Granot et al., 2007; Miller, 2007; Bahat et al., 2014). StAR is also recognized as a critical developmental factor for STAR gene expression organs. StAR is temporally and spatially expressed during the differentiation of the fetal adrenal cortex and gonads (Clark et al., 1994; Sugawara et al., 1995). Indeed, StAR expression coincides with key developmental milestones such as adrenal differentiation, gonadal formation, and gonadal sex differentiation (Hasegawa et al, 2000; Miller & Auchus, 2011; Clark & Stocco, 2014). Notably, classical disorders such as lipoid congenital adrenal hyperplasia (CAH) and nonclassic forms highlight the essential role of StAR. So far, extensive studies have been conducted to understand the mechanism and roles of StAR, and there are many good reviews (Manna & Stocco, 2005). In this review, we provide a retrospective overview of the general anatomy of the STAR gene and its biological functions.
STAR ASSOCIATED DEVELOPMENTAL DISORDERS AND OTHERS
The spatial and temporal precision of StAR expression reflects its indispensable role in orchestrating key events in endocrine organ formation and steroidogenesis (Merke & Bornstein, 2005; Miller, 2025). During early human development, StAR expression begins in the adrenal cortex around the sixth gestational week, shortly after the adrenal primordia emerge from the coelomic epithelium. In the gonads, particularly in 46, XY individuals, StAR is upregulated in fetal Leydig cells around the eighth to tenth week, coinciding with the initiation of testicular androgen production. In 46, XY fetuses, disrupted testosterone synthesis leads to incomplete virilization, failure of Wolffian duct maintenance, and persistence of Müllerian derivatives due to insufficient anti-Müllerian hormone (AMH) synergy. In 46, XX individuals, although fetal ovarian steroidogenesis is minimal, some reports suggest altered thecal cell development and aberrant folliculogenesis under conditions of chronic StAR deficiency. Similarly, adrenal hypoplasia, cortical disorganization, and defective capsule formation have been observed in both human patients and StAR-deficient mouse models.
Classical lipoid CAH is typically caused by null mutations in STAR (Lin et al., 1995; Bose et al., 1996). These mutations result in early-onset adrenal insufficiency, salt-wasting crises, and 46, XY sex reversal, due to failure of testosterone synthesis during the critical window for male genital development. Histological analyses of CAH fetal adrenal glands reveal the disruption of adrenocortical zonation and Leydig cell differentiation, leading to feminized external genitalia and regression of Wolffian structures despite the presence of testes (Sahakitrungruang et al., 2010). In contrast, nonclassic or partial lipoid CAH arises from hypomorphic missense mutations, often within the StAR-related lipid transfer (START) domain (e.g., R182L, L275P) (Bose et al., 2000). These mutations show normal genital development and neonatal adrenal function, but patients may exhibit delayed puberty, primary amenorrhea, or infertility, reflecting the persistent yet insufficient hormone production. This highlights a developmentally graded requirement for StAR: robust activity is essential during embryonic sex differentiation, while lower levels may suffice until postnatal hormonal reactivation.
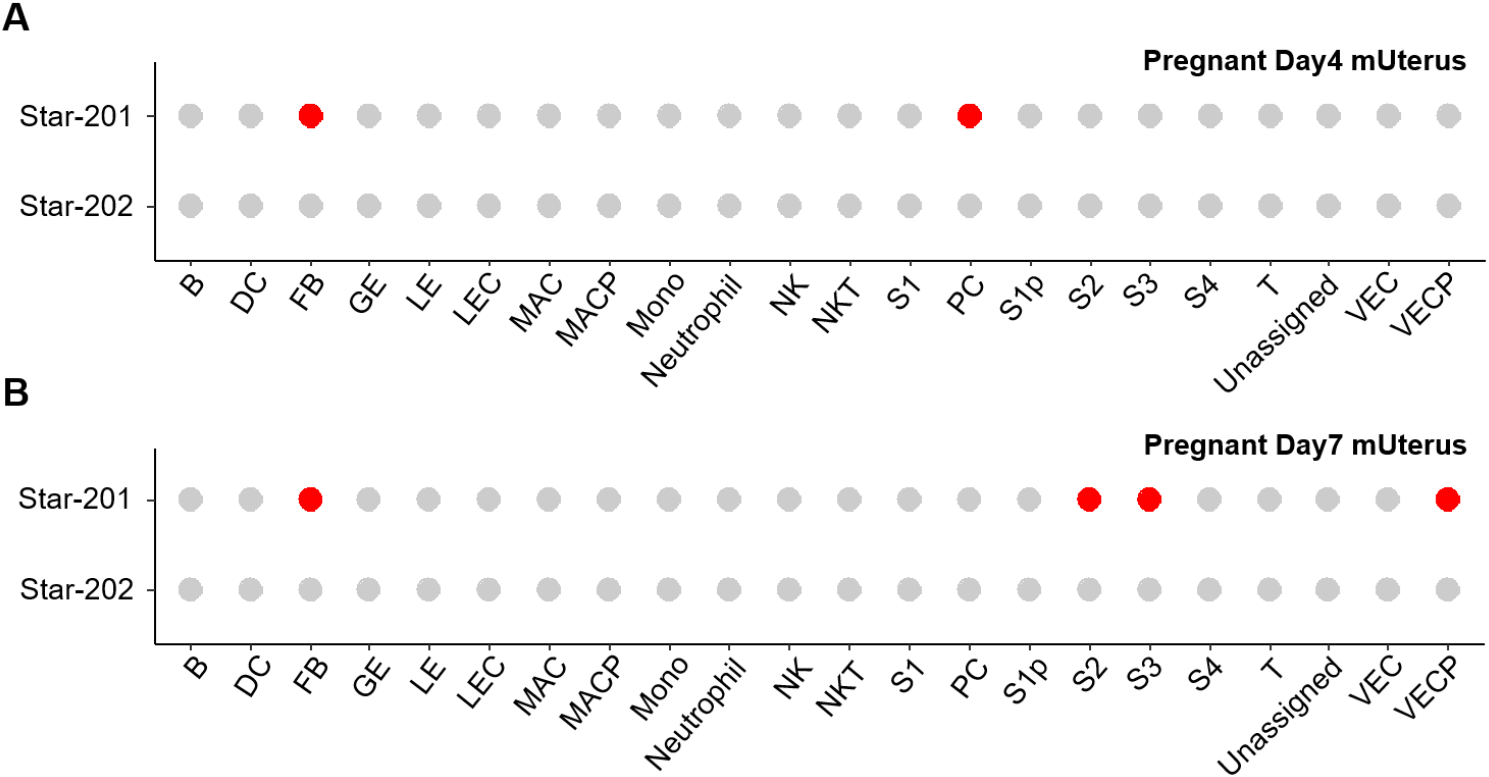
The adrenal glands and gonads are defective in StAR knockout model mice, and there are severe defects in corticosterone, adrenocortical insufficiency (Caron et al., 1997b). Both the absolute requirement for StAR during fetal adrenal and gonadal development and the spectrum of phenotypes observed illustrate how even subtle alterations in cholesterol transport can ripple through the architecture and function of developing endocrine systems. On the other hand, it is suggested that StAR could mediate the other protein function. The 30-kDa StAR facilitates the interaction of P450c11AS (11 beta-hydroxylase, aldosterone synthase) with Tom22 (mitochondrial import receptor subunit TOM22 homolog) (Bose et al., 2021). It is also revealed that the STAR isotype is expressed in differentiating stromal cells and proliferating vascular endothelial cells. Those were detected in decidualization but not in non-implanted uterine endometrium (unpublished) (Fig. 1). This suggests that StAR may be involved in cell proliferation and differentiation in tissues other than endocrine tissues, although further studies are needed. Those demonstrate that partial loss-of-function mutations in the STAR gene can have broad developmental impacts (Sahakitrungruang et al., 2010; Katharopoulos et al., 2020).
STAR GENE ANATOMY AND EXPRESSION REGULATION
The STAR gene is a protein-coding gene with synonym STARD1 in humans, and D8Ertd419e and STARD1 in mice (NCBI, 2025). The human STAR gene is located on chromosome 8p11.23 (ENSG00000147465; 38,142,700-38,150,992 reverse strand) and consists of 7 exons and 6 introns. A pseudo gene was mapped to chromosome 13 (Sugawara et al., 1995). The gene structure is highly conserved in mice, with the murine Star gene localized to chromosome 8 (8D3; ENSMUST00000033979.6) and consisting of 7 exons and 6 introns like in human (Clark et al., 1994). The exon-intron boundaries are nearly identical, indicating strong evolutionary conservation and supporting the relevance of mouse models in studying STAR gene function (Figs. 2 and 3).
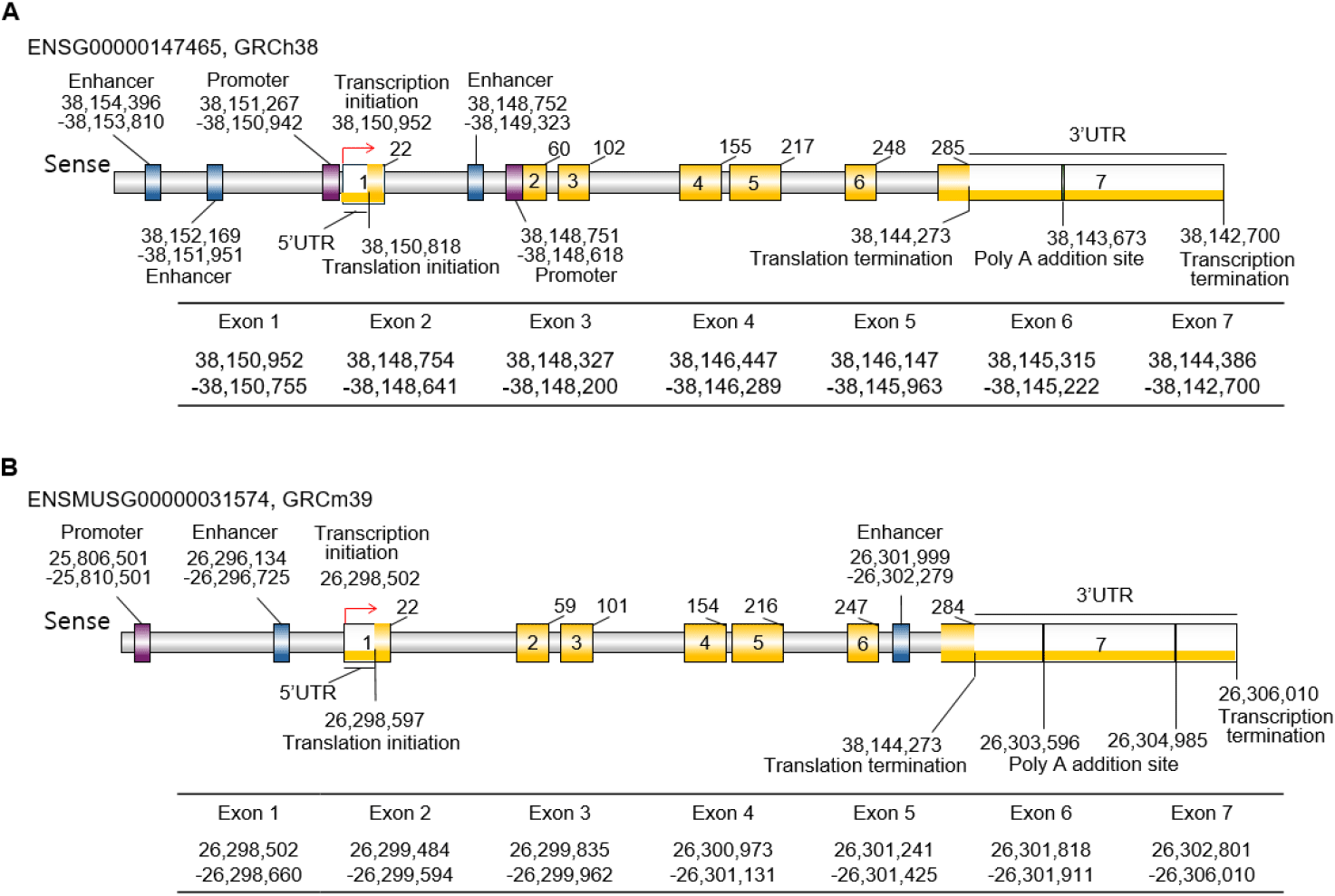

Notably, exon 1 includes the 5′untranslated region (UTR) and the initial portion of the mitochondrial leader sequence, which is continuous up to part of exon 3, while from that point onward, exons 3 through 7 encode the remaining portions and the functional core of the StAR protein. Exon 7 includes the stop codon, the 3’ UTR, and poly(A) addition sites. In humans, there is one PolyA signal sequence and one PolyA site, but in mice, 2 PolyA signal sequences and two PolyA sites, respectively (Figs. 2 and 3). These exons encode a 285 amino acid precursor protein in humans and a 284 amino acid one in mice (Figs. 2 and 3). The precursor protein includes a mitochondrial targeting sequence (MTS) at the N-terminus and a conserved START domain (exons 5 to 7) at the C-terminus (Clark et al., 1994; Sugawara et al., 1995; Arakane et al., 1996) (Figs. 2 and 3). The StARs are well conserved between species, and the similarity between human and mice is 87.32% (Fig. 3C).
TATA-box and CpG island are related to tissue specificity. TATA-box is thought to be absent in house-keeping genes but has a CpG island (Gardiner-Garden & Frommer, 1987; Zhu et al., 2008). Interestingly, they are not present in both human and mouse STAR genes (Fig. 3). The core promoter is localized in front of the exon 1 and exon 2, respectively, in human and just one promoter in mouse. The STAR promoter contains a lot of transcription factor (TF) binding sites, such as SF-1 and cAMP-response elements. Recently, Zhao et al. (2025) reported that there are multiple CpG regions but a lack of CpG islands at the core promoter of the STAR gene.
Generally, enhancers play crucial roles in spatiotemporal gene expression during development and are enriched for genetic variants that are associated with molecular and cellular traits (Javierre et al., 2016; GTEx Consortium, 2020; Chandra et al., 2021; Claringbould & Zaugg, 2021; Ray-Jones et al., 2025). The tissue-specific and developmental stage-specific expression of the STAR gene has been studied, and the utilization of enhancers to switch on or switch off gene expression is suggested. For example, STAR is expressed in the adrenal and the testis during development but not in the developing ovary (Clark et al., 1995). In the ovary, STAR expression is detected at puberty and at various ovulatory phases (Pollack et al., 1997; Ford et al., 1999). It is suggested that the cause of multiple abnormalities in adrenal and testis function of STAR knockout mice is developmental defects of these steroidogenic organs (Caron et al., 1997a,b). So far, the TFs binding to the motifs in the promoter, SF-1 and DAX-1, are known as factors for spatio-temporal regulation. However, it is hard to find the published data for the possible roles of enhancer, and the possible roles in tissue and developmental stage specificity of enhancer should be evaluated. From the preliminary analysis of our laboratory, the enhancers are expected in the mouse. Besides, based on the Ensembl (Dyer et al., 2025), there are enhancers. In humans, there are 2 enhancers in front of the promoter, and 1 is in front of exon 2. In mice, there are a total of 2 enhancers, one is located in front of exon 1, and the other is in exon 7. The STAR gene has an enhancer site both in human and mouse at intron 1 and 2, and intron 6, respectively (Fig. 2). The enhancer of the human STAR gene has 88 TF binding motifs and 51 motifs in mice (FIMO: Version 5.5.8 compiled on May 17 2025 at 17:04:22, https://meme-suite.org/meme/doc/fimo-output-format.html#tsv_results).
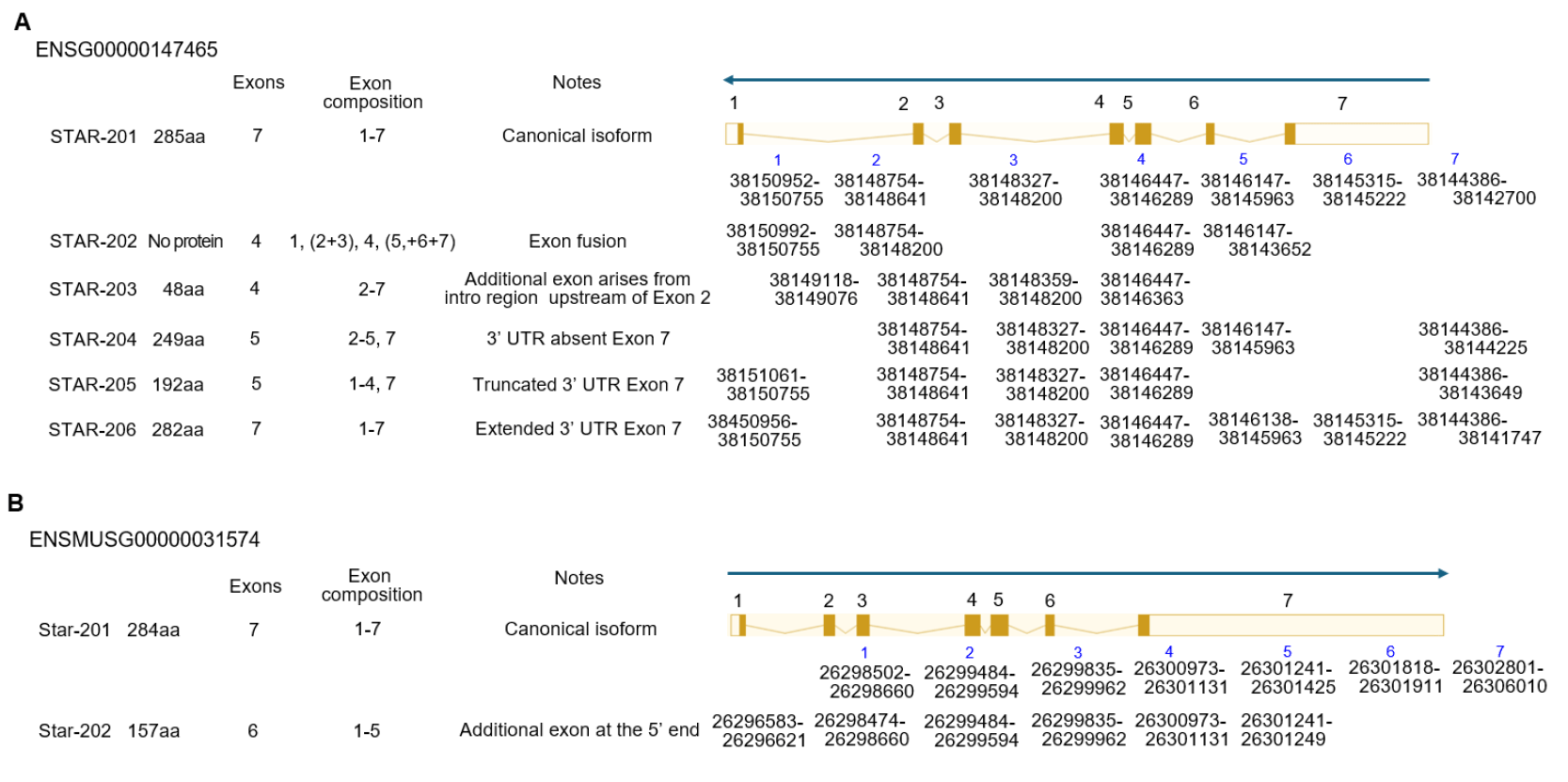
Alternative splicing of STAR transcripts is typically transcribed as a single dominant START isoform in steroidogenic tissues. So far, 6 spliced mRNAs (STAR-201, STAR-202, STAR-203, STAR-204, STAR-205, and STAR-206) have been evaluated, and one of them, STAR-202, does not make protein in the human STAR gene. In the case of mice, there are 2 spliced forms, STAR-201 and STAR-202, in Ensembl (Dyer et al., 2025; Vo et al., 2025) (Fig. 4). In pregnant day 4 uterine endometrium, only the STAR-201 isoform is detected at fibroblast and PC. Interestingly, this isotype was detected in fibroblast and differentiating stromal cells, but not in PC in decidualized tissue (unpublished) (Fig. 1).
Transcription of the STAR gene is tightly regulated by both tissue-specific and hormone-responsive mechanisms that ensure precise temporal and spatial expression in steroidogenic tissues (Clark et al., 1994; Sugawara et al., 1995; Luo et al., 1998; Miller & Bose, 2011; Rawan et al., 2023). One of the interesting things is its rapid induction in response to hormones such as adrenal corticotropic hormone (ACTH), luteinizing hormone (LH), growth hormone (GH), angiotensin II, or retinoic acids. Hypoxia and circadian clocks are also known factors of expression regulation of STAR (Li et al., 2003; Son et al., 2008; Kowalewski, et al., 2015; Wang et al., 2019; Baddela et al., 2020; Xiao et al., 2021). In addition to the increasing stimulation for STAR mRNA levels, the LPS, Heat shock, TGFβ, PGF2α and IFγ are known as factors for lower STAR mRNA levels (Reinhart et al., 1999). Most of them use the second messenger cAMP. cAMP-mediated steroidogenesis is a well-known for StAR expression involved in the cAMP-PKA axis (Manna et al., 2002). The other suggested mediators are calcium, arachidonic acids, chloride ion channels, and mitogen-activated protein kinase/extracellular signal-related kinase. Ca2+ involves SF-1 expression through Ca2+/calmodulin-dependent protein kinase 1 (CAMK1)-dependent pathway (Manna & Stocco, 2005; Martin & Tremblay, 2008).
As mentioned previously, these mechanisms are mediated through epigenetic factors such as chromatin topologies and TFs interacting within the promoter. Tissue or developmental-stage-specific expression of a gene depends on the history of the cell lineage, epigenetic memory. Epigenetic regulation of the STAR gene expression has also been explored recently. Along with increased histone acetylation at the locus with DNA methylation dynamics, ChIP studies have shown direct binding of SF-1, CREB, and CBP/p300 to the endogenous STAR promoter following cAMP stimulation (Manna et al., 2002). YY1-mediated histone deacetylation is identified in adrenal cells in response to nicotine, a known repressor of fetal adrenal STAR expression (Liu et al., 2016). A large number of TFs that interact with a tightly clustered promoter region over a relatively short promoter region is involved in the complexity of STAR gene transcription. STAR gene promoter contains GC-rich sequences and cAMP response elements (CREs) (Sugawara et al., 1997). In addition, about 44 TFs are identified, such as NR5A1, NROB1, GATA4, DLX5, DLX6, FOXL2, FOXO3, JUN, CREB, YY1, PAX6, etc. (Viger et al., 2024). From such overrepresented TF binding sites, the kinds of TF binding motifs, and TATA box and rich CpG sequences in the promoter, it is suggested that the STAR could work as an immediate-early gene (unpublished data; based on the criteria for immediate early gene) (Bahrami & Drabløs, 2016). Although the immediate early gene, NR4A is an important (TF) for StAR expression.
The extensive studies evaluated the role of TFs in STAR gene expression. The NR5A1 (also known steroidogenic factor-1, SF-1), a nuclear receptor that binds to specific response elements in the proximal promoter of the STAR gene with variability by species and cell context, is a key transcriptional regulator (Clark et al., 1995; Sugawara et al., 2000; Wickenheisser et al., 2000; Hiroi et al., 2004). It is essential for both basal transcription and cAMP-stimulated expression of STAR. Deletion or mutation of NR5A1 motif in promoter significantly reduces STAR promoter activity. NR5A1 interacts with a few TFs to either upregulate or downregulate STAR gene transcription. CBP/p300, transcriptional coactivators with histone acetyltransferase activity, are recruited by both NR5A1 and phosphorylated CREB, facilitating chromatin remodeling and transcriptional initiation (Manna et al., 2002; Sewer & Waterman, 2003). CREB-related family members, CREB, cAMP response element modulator (CREM), and ATF1, are implicated in STAR gene expression in the major steroidogenic tissues (Lanfranchi et al., 2022).
NR0B1 TF (also known as dosage sensitive sex reversal adrenal hypoplasia critical region, on chromosome X, gene 1 or DAX1) binds directly to the DNA hairpin structures on the STAR promoter (Manna et al., 2009a). It prevents the transcriptional activity of NR5A1 in STAR as a negative coregulator and functions as a negative sensitizer of hormone/cAMP-induced STAR gene expression in steroidogenic cells (Iyer & McCabe, 2004; Pandey et al., 2010; Li et al., 2011). On the other hand, another TFs such as CCAAT enhancer binding protein beta (CEBPB) (Reinhart et al., 1999), specificity protein 1 (Sugawara et al., 2004), activator protein 1 (AP1) TF subunits JUN, FOS, and CRE-activating transcription factor 2 (ATF2) (Manna et al., 2004; Pierre & Tremblay, 2022), sterol regulatory element binding TF (Shea-Eaton et al., 2001), CEBPB (Silverman et al., 1999), GATA4 (Tremblay et al., 2002), SP1 (Sugawara et al., 2000), SREBF1 (Shea-Eaton et al., 2001) are cooperative partner of NR5A1 for STAR gene expression (Tremblay & Viger, 2001; Manna et al., 2009b). NR4A1, a member of NR4A family, is an alternative candidate TF over NR5A1 for STAR transcription in steroidogenic cells (Abdou et al., 2013; Zhang et al., 2013). GATA4 and GATA6 TF are suggested as an essential TF for STAR expression regulation (Bouchard et al., 2022). GATA factors interact with various TFs (for example CEBPB, JUN, MEF2, FOS and FOG2) to either positively or negatively modulate STAR promoter activity in steroidogenic cell lines (Tremblay et al., 2002; Tevosian et al., 2015; Bouchard et al., 2022). JUN and CREB can activate STAR transcription but FOS suppress that’s expression (Manna & Stocco, 2007). It also suggested that GATA may be involved in the acute increase in STAR transcription through cAMP stimulation (Hiroi et al., 2004; Silverman et al., 2006).
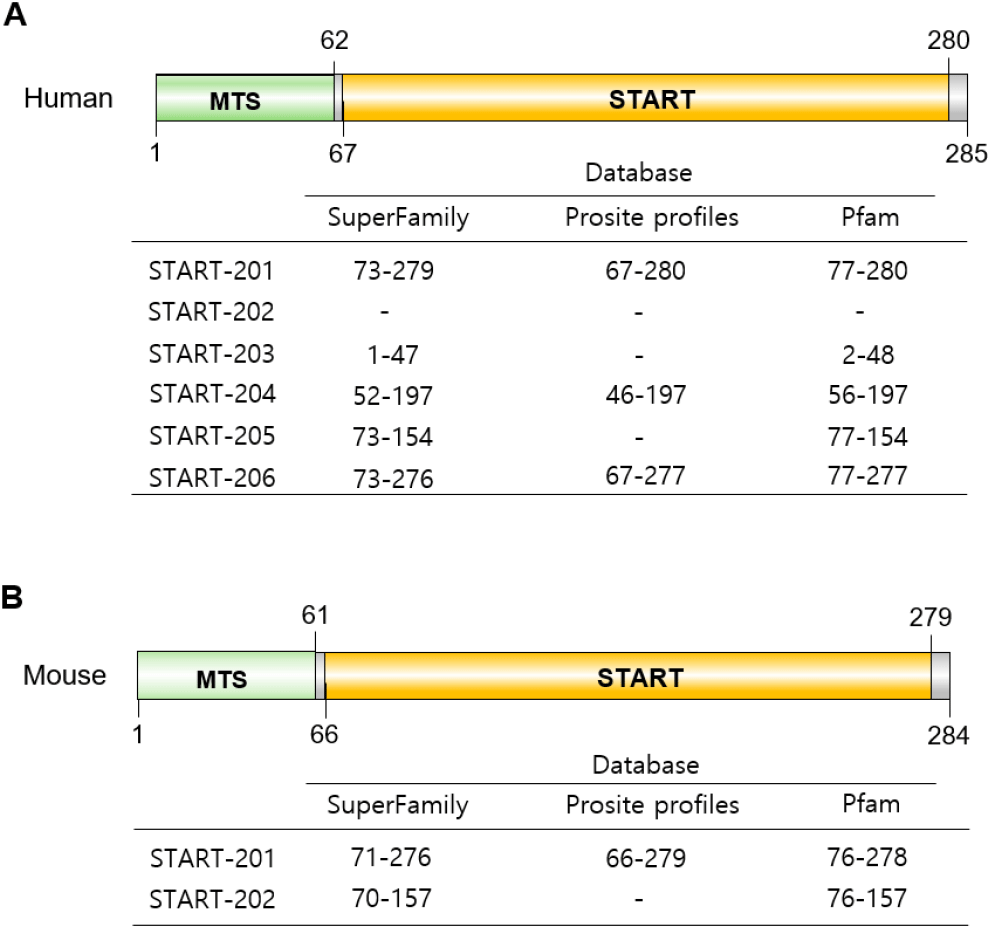
StAR belongs to a family of structurally related proteins that contains the START (Tugaeva & Sluchanko, 2019). It is known as two major distinct regions in STAR protein; one is START domain and the other is the MTS (Christenson & Strauss, 2000). In humans, the N-terminal mitochondrial sequence is from 1 to 62, and in mice, from 1 to 61 (Arakane et al., 1996) (Fig. 5). MTS directs it to the mitochondria and is located in the N-terminal domain of the murine and human StAR proteins. MTS interacts with its receptors on the mitochondrial surface, such as the translocase of outer membrane (TOM) complex (Neve & Ingelman-Sundberg, 2001), and there are cleavage sites for mitochondrial protease. The START domain serves as the primary cholesterol-binding module. In START family, the START domains conserve–210 amino acids (range from 200 to 210 amino acids) in length and adopts a conserved α/β helix-grip fold that reates a deep hydrophobic cavity suitable for binding cholesterol and other lipids such as oxysterols, phospholipids, sphingolipids, and fatty acids (Kallen et al., 1998; Ponting & Aravind, 1999; Clark, 2012). The START domain is located at 67-280 in human and 66-279 in mouse StAR proteins. This domain is not only critical for StAR’s cholesterol-binding capacity but is also required for its proper structural flexibility and membrane interaction (Fig. 5). StAR protein (from STAR-201 mRNA) is synthesized as a 37 kDa active cytosolic protein, and upon hormonal stimulation, it moves to the outer mitochondrial membrane (OMM), where it functions in steroidogenesis and where its MTS is cleaved, yielding an inactive 30 kDa protein: 37 kDa cytoplasmic form with a short half-life of about 15 min and 30 kDa intramitochondrial form with several hours (Miller, 2007).
The activity of the StAR protein involves multiple posttranslational regulatory mechanisms that ensure its acute responsiveness and spatial specificity in steroidogenic cells. Among these, phosphorylation plays a key role. StAR is phosphorylated at human Ser195 (Ser194 in mouse) by protein kinase A (PKA) through type II PKA regulatory subunit (PKAR2A) by the PKA anchor protein AKAP121. Such phosphorylation enhances about 2 times its cholesterol transport activity through conformational change or the interaction with other mitochondrial proteins (Arakane et al., 1997; Stocco et al., 2005). Importantly, substitution of this residue with alanine markedly reduces StAR function; knock-in of S194A StAR does not rescue embryonic lethality, but not in WT StAR knock-in (Sasaki et al., 2014). Each of 30 kDa or 37 kDa could be phosphorylated forms or not (Miller, 2007).
A unique aspect of StAR’s posttranslational regulation is its non-classical folding behavior. Unlike many fully folded globular proteins, StAR appears to adopt a molten globule-like intermediate state, characterized by partial secondary structure and conformational flexibility. This state is hypothesized to be critical for membrane association and lipid transfer. Biophysical assays, including limited proteolysis, fluorescence, and circular dichroism have shown that StAR remains structurally dynamic in solution and gains functional conformation upon interaction with the mitochondrial membrane (Arakane et al., 1998; Bose et al., 1999). This unusual folding pattern may allow StAR to transiently bind cholesterol and deliver it across the OMM to the inner mitochondrial membrane (IMM) enzyme CYP11A1 (Bose et al., 1999; Miller, 2007).
The rapid turnover of StAR reflects its tightly regulated function. It has a short half-life, typically less than 1 hour, and is rapidly degraded in the absence of successful mitochondrial import. Cytosolic StAR is cleared by both proteasomal pathways and mitochondrial quality control mechanisms, such as Lon protease-mediated degradation, which prevent the accumulation of mislocalized or nonfunctional protein (Granot et al., 2007). This ensures that StAR activity is both temporally limited and spatially precise, aligning with the episodic nature of steroid hormone production.
MECHANISM OF STAR PROTEIN IN CHOLESTEROL TRANSPORT
The StAR protein is modulated not only by its structural conformation and mitochondrial targeting but also by its interactions with other mitochondrial proteins that participate in cholesterol transport. The N-terminal MTS facilitates the delivery of StAR to the OMM (Clark et al., 1994; Arakane et al., 1997). StAR acts at the OMM as a soluble protein and promotes cholesterol movement through a non-vesicular, proteinmediated mechanism (Arakane et al., 1997; Miller, 2007). This rapid translocation process does not require complete mitochondrial import of the protein, unlike classical mitochondrial matrix proteins (Lin et al., 1995; Granot et al., 2007). Determining the activity of StAR is proportional to its residency time on the OMM and StAR’s cellular localization, and not cleavage from 37 to 30 kDa.
While structurally similar domains exist in proteins such as StarD3 (MLN64) and StarD4, only StAR exhibits rapid, hormonally responsive activity in steroidogenic tissues. This is likely due to the integration of its conformational flexibility with acute mitochondrial targeting features that distinguish StAR from its paralogs (Christenson & Strauss, 2001; Murcia et al., 2006). Upon membrane contact, conformational changes stabilize the cholesterolbinding cavity within the START domain, enhancing transfer to down-stream mitochondrial acceptors (Bose et al., 2000). Molecular dynamics simulations and mutagenesis analyses have shown that disruption of conformational mobility within the START domain—particularly near the C-terminal α-helix—impairs cholesterol transfer without affecting mitochondrial localization, underscoring that structure-function dynamics within the domain are essential for activity (Arakane et al., 1998; Miller, 2007). Importantly, over 95% of known pathogenic missense mutations in the STAR gene—including E169G, R182L, and L275P—are clustered within the START domain (Lin et al., 1995; Sahakitrungruang et al., 2010). These mutations are typically located in or near the cholesterol-binding cavity and are thought to impair either ligand affinity or the conformational changes required for lipid transfer. Dysfunction of StAR results in clinical manifestations of lipoid CAH.
Mechanistically, StAR is thought to operate as part of a multiprotein assembly known as the transduceosome, a cholesterol transfer complex assembled at OMM–IMM contact sites. Transduceosome includes the voltage-dependent anion channel (VDAC-1), the translocator protein (TSPO), TSPO-associated protein 7 (PAP7, ACBD3 for acyl-CoA-binding-domain 3), ATPase family AAA domain containing 3A (ATAD3A), kinase A regulatory subunit 1α (PKAR1A), and other accessory proteins, which together may form a scaffold to relay cholesterol to the IMM (Papadopoulos et al., 2006; Rone et al., 2012). In addition, other regulatory molecules including heat shock proteins (HSPs) and cytoskeletal elements-have been suggested to indirectly influence StAR function by affecting its folding, trafficking, or delivery to the mitochondria (Bose et al., 2000). Although the exact molecular interactions remain under investigation, experimental evidence supports close proximity and collaboration between StAR and these components during steroidogenic stimulation.
Mounting evidence supports a model in which StAR must remain at the OMM long enough to complete cholesterol transfer. Studies have shown that delaying the mitochondrial import of StAR—such as through mutations that interfere with import kinetics—can increase steroidogenic output, while rapid import correlates with reduced activity (Watari et al., 1997; Miller, 2025). Once fully imported into the mitochondrial matrix or inner membrane space, StAR is subjected to proteolytic degradation by Lon protease, effectively terminating its activity (Bahat et al., 2014). These findings suggest that StAR’s active state is temporally restricted to its membrane associated, preimport phase.
OMM and IMM microstructure also involved in efficient cholesterol transport. The microstructure formed through steroidogenic OMM/IMM contact, may be involved in cholesterol transport. 67-kDa protein ATAD3 is anchored at mitochondrial contacts site and facilitates contact between the IMM and the OMM (Gilquin et al., 2010). The microstructure permits a gross molar transport with the conformation change of StAR (Rajapaksha et al., 2013). In addition, the mitochondria-associated membrane (MAM), the interaction site between mitochondria and ER, is concerned in cholesterol transport to mitochondria (Vance, 2014).
The TOM complex, import channel, is a known for cholesterol-StAR transport (Bose et al., 2021, 2023). After transport cholesterol, the StAR is degraded by Lon protease (Bahat et al., 2014). It is generally accepted that more studies are needed, although many studies have been done. Because it is still not clear how StAR facilitates the rapid and massive influx of cholesterol (Miller, 2025).
CONCLUSION
The two-hit model of lipoid CAH can be deduced from StAR-dependent or -independent steroidogenesis. Non-steroidogenic cells transfected with the side chain cleavage system (P450scc/FDX/FDXR) synthesize pregnenolone in the absence of StAR at about 14% of the StAR-induced rate (Lin et al., 1995). A few molecules, START domain (conserved sequence of ~210 amino acids) proteins (15 members, StarD1-StarD15; STAR is STARD1, MLN64 is StarD3) are suggested as functionally redundant molecules. A transient structurally adaptive mechanism that integrates dynamic protein folding, membrane association, and coordinated lipid binding causes the cholesterol transport of StAR. This finely tuned process is indispensable for steroid biosynthesis and represents a specialized mode of intracellular lipid trafficking in mammalian endocrine systems.
Beyond its biochemical function, StAR has emerged as a key developmental regulator, with its expression precisely timed and spatially restricted to steroidogenic tissues during embryogenesis (Clark et al., 1994; Sugawara et al., 1995). This spatiotemporal regulation ensures that steroid hormone production coincides with critical windows of adrenal and gonadal organogenesis, enabling proper sexual differentiation, endocrine tissue morphogenesis, and homeostatic programming (Sahakitrungruang et al., 2010).
The precise molecular mechanism by which StAR facilitates cholesterol transfer, the identity and coordination of its interacting partners, and the developmental cues governing its expression still require further elucidation (Papadopoulos et al., 2006; Rone et al., 2012). Advances in stem cell models, gene editing technologies, and in vivo imaging hold promise for uncovering these unknowns (Ruiz-Babot et al., 2023). As steroidogenic disorders continue to be diagnosed with increasing genetic precision, understanding the developmental framework of StAR function becomes not only a matter of academic interest but also a foundation for therapeutic insight. Clarifying the role of StAR in embryonic endocrine development may pave the way for novel interventions, including targeted gene therapies or strategies for functional rescue in hypomorphic conditions. Ultimately, the study of StAR offers a unique lens through which to explore the interplay between molecular transport, cellular differentiation, and the developmental origins of endocrine health.