INTRODUCTION
Pluripotent embryonic stem (ES) cells, which are mainly used in chimeric mice production for knockout mice studies, are derived from the inner cell mass (ICM)/epiblast population of the blastocyst (Johnson & Ziomek, 1981). Although the ICM is a transitory cell population in the embryo, cultured ES cells can possess an unlimited self-renewal capability and maintain the pluripotent state. From the studies of Thomas & Capecchi (1987) that demonstrated that any genes could be mutated by homologous recombination in ES cells, mouse embryonic stem cells (mESCs) have been used for producing chimeric embryos to make genetically modified mice. However, due to the unstable epigenetic states in mESCs during manipulation in vitro (Humpherys et al., 2001), many efforts for sustaining stable states during long-term culture have recently been reported (Ramirez et al., 2009).
In mammals, many epigenetic modifications (e.g., DNA methylation, histone acetylation and methylation to maintain the pluripotency of ES cells) have been studied, and their effect on development and differentiation has been characterized. DNA methylation at cytosine in the CpG island of differentiation marker genes and histone lysine acetylation of polycomb group complexes to maintain the pluripotency of ES cells are presently best understood (Boyer et al., 2006; Ha et al., 2011; Leeb & Wutz, 2012). Moreover, histone methylation of lysine residues has been highlighted as studies of histone modifications but not arginine residues. However, recently, the importance of histone arginine methylation has been focused on the epigenetic regulation of pluripotency in ES cells (Wu et al., 2009).
The histone methylation of arginine residue as a histone modification is catalyzed by the protein arginine methyl-transferase (PRMT) family, which consists of 10 members in mammalian cells (Bedford, 2007). Among the PRMT family, coactivator-associated arginine methyltransferase 1 (CARM1)/PRMT4 is known to catalyze the asymmetric di-methylation of the arginine residues of many proteins, such as histones and certain transcription factors, and the enzyme acts as a coactivator for transactivation (Bedford, 2007). CARM1 functions as a secondary coactivator with the p160 transcriptional coactivator family which includes SRC-1, GRIP1/TIF2, and p/CIP and mediates transcriptional activation by nuclear hormone receptors (Chen et al., 1999). In addition, CARM1 can methylate histone H3, and the coactivator-mediated methylation of proteins in the transcription machinery may contribute to transcriptional regulation (Feng et al., 2006).
As histone H3 methyltransferase, CARM1 methylates histone H3 arginine 17 and 26 residues that recently emerged as an epigenetic mechanism regulating pluripotency in mouse embryos (Torres-Padilla et al., 2007). The elevation of the expression of CARM1 in individual blastomeres leads to increased histone methylation and results in higher expression of pluripotency transcription factors such as Oct3/4, Nanog, and Sox2 and direction of progeny to the ICM (Torres-Padilla et al., 2007). Even though all cells are initially pluripotent, reduced CARM1 activity predisposes cells to differentiate (Wu et al., 2009).
As an easy, efficient, and precise method of intracytoplasmic delivery of CARM1 protein, we used a cell-penetrating peptide (CPP)-mediated protein-delivery system (Jo et al., 2010). In Jo et al. (2012), when the CPP-CARM1 protein was delivered exogenously into human mesenchymal stem cells (MSCs), it was able to methylate transiently histone H3 arginine residues, and it resulted in the up-regulation of pluripotency-related genes and differentiation potentials. In the present study, to increase the efficiency of the production of chimeric mice and germ-line transmission, we produced a mouse recombinant CARM1 protein and analyzed its function after intracytoplasmic delivery into mESCs.
MATERIAL AND METHODS
The CARM1 cDNA clone was purchased from Imagenes (I.M.A.G.E full length cDNA clone; Imagenes, Berlin, Germany) and it was used as templates for polymerase chain reaction (PCR). CARM1 cDNA were amplified including restriction enzyme sequences at 5’-Xho I (CTCGAC) and start codon, 3’-BamHI (GGATCC). The PCR cycle conditions were 95°C for 30 s, 60°C for 1 min, 72°C for 1 min (30 cycles). These PCR products were inserted into the pCR®8/GW/TOPO vector using pCR®8/GW/TOPO® TA Cloning Kit (Invitrogen, Carlsbad, CA) and confirmed by gene sequencing. CARM1 ORF was replaced into pET-20b vector which includes 7x oligoarginine (7R) as a CPP by Xho I and BamHI restriction enzyme.
The constructed protein expression vector, CPP-conjugated CARM1 (CPP-CARM1), were transformed into BL21(DE3)pLysS competent cells (Stratagene Inc., La Jolla, CA, http;//www.stratagene.com). The cells were cultured on LB-agar plate containing 100 μg/ml ampicillin (Sigma Aldrich, St. Lois, MO). After that, the desired single colonies were inoculated into 3 ml of LB medium containing 100 μg/ml ampicillin and grown for 16 hr in 37°C shaking incubator. The grown cells were large-cultured as 1:100 ratio in 200 ml of LB medium for 2 hr without antibiotics (OD 600 : 0.8). Induction were performed by 1 mM IPTG (Isopropyl-1-thio-β-D galactopyranoside : Sigma Aldrich) for 8 hr at 20°C, then pellet were collected by centrifugation. NP-10 [50 mM NaH2PO4 (Sigma Aldrich), 300 mM NaCl (Sigma Aldrich), 10 mM imidazole (Sigma Aldrich), pH to 8.0] and lysozyme (Sigma Aldrich) solution (10 mg/ml) 1 ml (10 mg), and Benzonase (3 units/ml : QIAGEN, Inc.) were added into collected cells and stood for 30 min at room temperature for lysis. Then, lysate pellet was centrifuged at 15,000 rpm and separated supernatant were collected. Soluble supernatant were purified by His-Tagged Recombinant Protein Purification through Ni-NTA Superflow Columns (QIAGEN Inc.).
Undifferentiated mouse ES cells, which originated from GFP expressing transgenic mice [C57BL/6-Tg(CAG- EGFP), Japan SLC. Inc] were cultured on irradiated CF1 mouse embryonic fibroblasts (MEFs) in 6-well plates (Nunc, Roskilde, Denmark) coated with 0.1% gelatin (Sigma Aldrich). Sub-culturing was performed every 3–4 days onto new MEF-seeded dishes by 0.05% Trypsin-EDTA (Hyclone, Logan, UT) for 3min. Cells were maintained in ES medium that consisted of Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (Hyclone) and supplemented with 20% knockout serum replacement (KSR, Gibco-BRL Frankin Lakes, NJ), 1% (v/v) non-essential amino acid (Gibco-BRL), 50 μl β-mercapto ethanol (Gibco-BRL), 100 U/ml penicillin G, 100 μg/ml streptomycin (Gibco-BRL) and 103 U/ml ESGRO (LIF, Chemicon, Temecula, CA). The mouse ES cells were cultured at 37°C in 5% humidified CO2 incubator. On the next day after subculture, media was changed with or without 2 μg/ml CPP-CARM1. Twenty-four hours later, media was replaced in same pattern. Total CPP-CARM1 treatment was performed twice.
Mouse ES cells were cultured with or without CPP-CARM1 for 24 hours were fixed in 4% paraformaldehyde (PFA) at room temperature for 30 min, washed twice with PBS and permeabilized with 0.1% Triton X-100 (Sigma Aldrich) for 15 min. Cells were blocked in protein block solution (Dako North America, Inc., CA) for 1 hour at room temperature, and stained with primary antibodies anti-His-taq (Sigma, 1:100) and anti-H3R17di-me (Novus, 1:100), followed by 1 hour incubation a room temperature with Alexa Fluor 555-labeled goat anti-rabbit secondary antibodies and Alexa Fluor 546-labed anti-mouse secondary antibodies (Molecular Probes, Eugene, OR) diluted to 1:500 with DPBS for 1 hour at room temperature. All samples were counterstained with 1 μg/ml 4,6-diamidino 2-phenyindiol (DAPI; Sigma Aldrich) diluted to 1:1000 with DPBS for 15 min at room temperature and mounted using Vector Shield mounting medium (Vector laboratories, Inc., Burlingame, CA). All sample images were captured with a Carl Zeiss LSM 510 META confocal laser-scanning microscope (Zeiss, Jena, Germany). The stained cells images were reconstructed using LSM 510 META software. Table 1.
Purified CPP-CARM1 recombinant protein was separated by 8% sodium dodecyl sulphate-polyacrylamide gel electrophoresis, and they were transferred onto polyvinylidene difluoride membranes (PVDF, Bio-Rad Laboratories, Hercules, CA). The membrane was blocked in 3% BSA in DPBS contained 0.1% Tween-20 (DPBST, Sigma Aldrich, St Louis, MO), and incubated with primary antibodies (anti His-taq mouse IgG : Sigma Aldrich) at 1:1000 dilution in DPBST. Proteins were detected with horseradish peroxidase-conjugated secondary antibody (Bio-Rad) at 1:2000 dilution in DPBS for 1 hour. Immunoreactivity was detected using Western Blotting Luminol Reagent (Santa Cruz Biotechnology, Santa Cruz, CA).
Total cellular RNA was extracted from cultured cells using the TRIzol Reagent (Invitrogen), and each sample were quantified using Thermo Scientific NanoDrop™ 2000 Spectrophotometer. First strand cDNA was synthesized using PrimeScript 1st strand cDNA Synthesis kit (TakaraBio, Shiga, Japan) according to manufacturer’s instructions. Reverse transcription PCR was conducted using G-taq kit (Cosmo, Seoul, South Korea). All PCR products were separated by 2% agarose-gel electrophoresis. For quantification of expression level, real-time PCR was performed by using iQ™ SYBR® Green Supermix (Bio-rad Laboratories, Hercules, CA) on BIO-RAD CFX96™ real-Time System. The delta-delta Ct method was applied to normalize the expression levels of pluripotency-related genes and differentiation marker genes to those of 18S and β-actin as normalization control.
RESULTS
We constructed a vector for protein production. Mouse CARM1 cDNA sequences (1,577 bp) were obtained by PCR, and the cDNA clones were transferred into a pET-20b expression vector. A modified pET-20b vector was designed with the sequence of R7, which has a role in CPP at the end of the 5’ of the CARM1 protein-coding sequences and the histidine coding sequence at the end of the 3’ of the CARM1 coding sequences to facilitate purifying proteins (Fig. 1A). The accurate ORF was confirmed by sequencing analysis using a T7 promoter and T7 terminator primers.
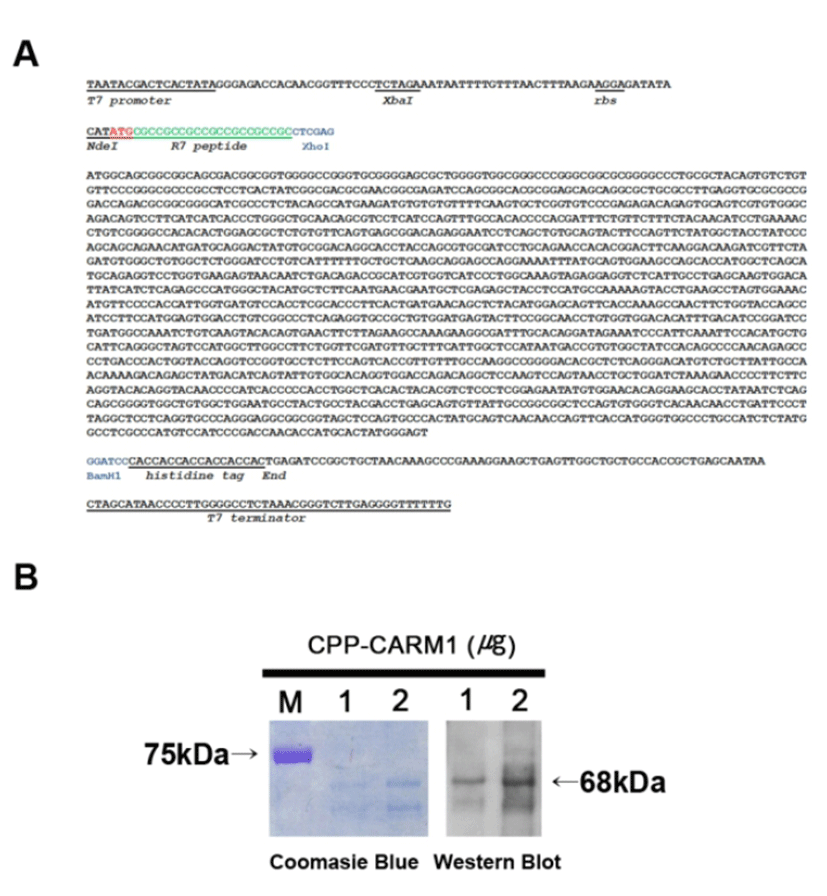
The CPP-CARM1 protein expression vector that was transformed into BL21(DE3)pLysS was induced to express protein by 1 mM IPTG and purified using 6x histidine-tag at the C-terminal of the protein. Soluble forms of CPP-CARM1 were obtained by culture at 20°C, and electrophoresis was performed by SDS-PAGE and confirmed by Coomassie Brilliant Blue Staining. In the process of purifying the protein, urea was not used for the preservation of protein activity, and the protein buffer was changed to DPBS containing 10% glycerol for minimal cytotoxicity. Purified CPP-CARM1 (1 and 2 μg) was confirmed by Western blot (Fig. 1B). As a result, the 68kDa CPP-CARM1 band was successfully detected.
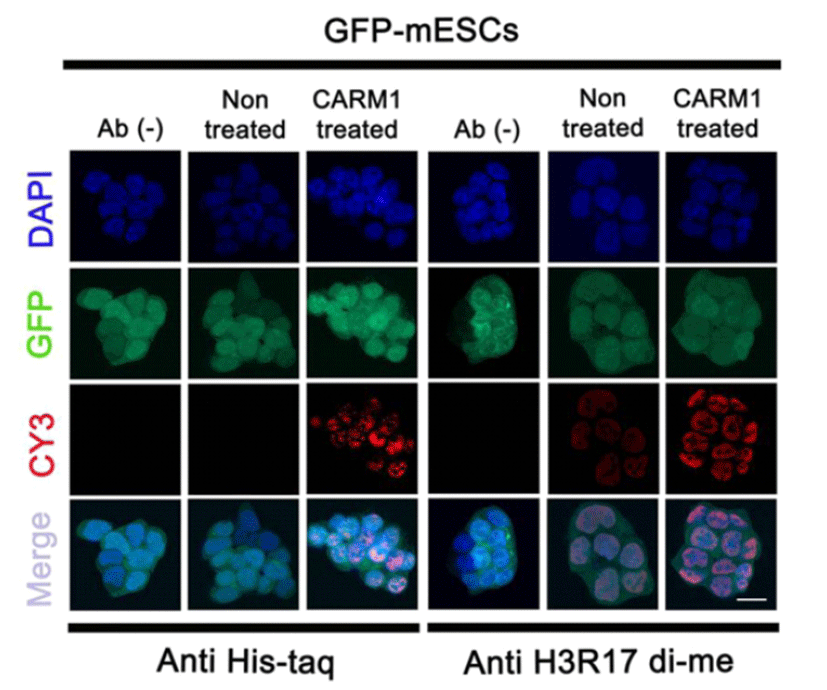
Based on a previous study in which CPP-CARM1 was observed inside the nucleus within 24 hr. (Jo et al., 2012), the 2 μg/ml CPP-CARM1 was treated into mouse ES medium. To check the translocation of the protein into the nuclei of the mESCs, cells were fixed at the time of the 24 hr. treatment and immunostained with anti-Histag antibody. The CPP-CARM1 was located in both the cytoplasm and nuclei. The recombinant protein was detected in most of the nuclei of the mESCs. In addition, immunostaining using anti-H3R17di-me antibody precisely showed the function of CARM1 at the histone H3 arginine 17 residue (Fig. 2). Therefore, similar to the previous study with human MSCs using CPP-CARM1, these results verified the translocation and function of CPP-CARM1 in mESCs.
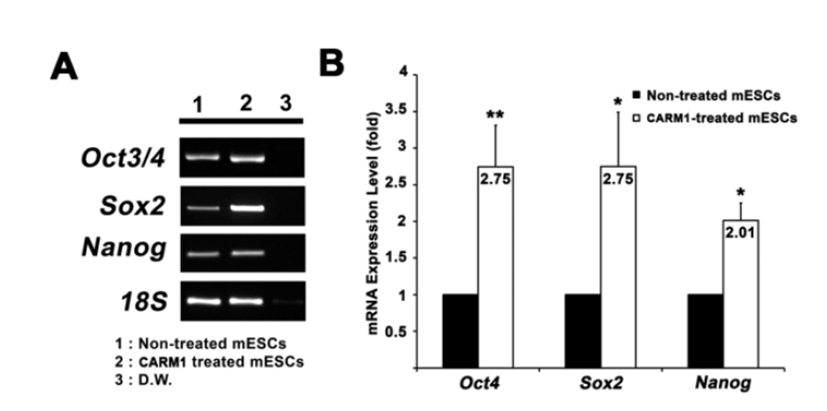
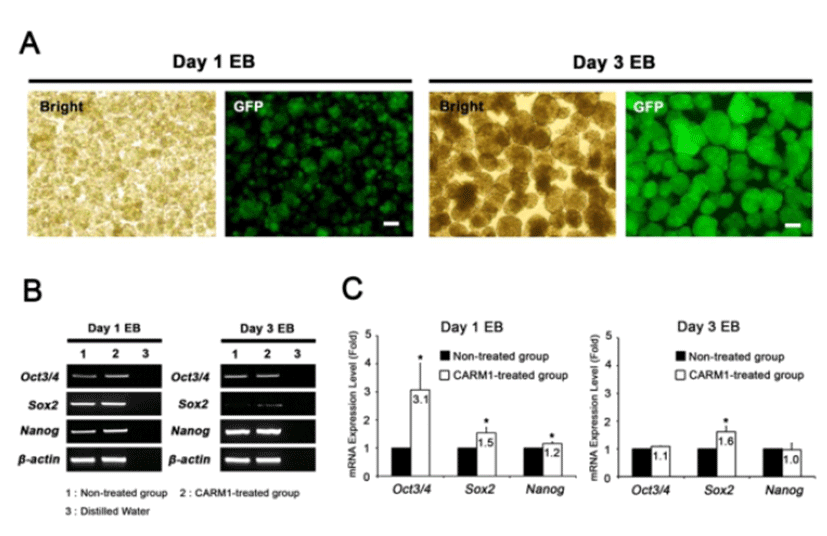
CARM1 associates with Oct4 and Sox2 promoters in human MSCs and regulates their expression levels (Jo et al., 2012). After treatment with or without CPP-CARM1 protein, the Oct4, Sox2, and Nanog expression levels of mESCs from the non-treated group and the protein-treated group were compared by CPP-CARM1 treatment led to an approximately two-fold increase in the of pluripotency-related genes compared to the control group (Fig. 3). In addition, after treatment with CPP-CARM1, spontaneous differentiation through the formation of EBs occurred in culture media without, but this was delayed compared to the non-treated group. In other words, the pluripotency-related gene expression level of CPP-CARM1-treated mESCs was decreased slightly when spontaneous differentiation was induced for 1 day but not markedly distinguished for 3 days of differentiation when compared with the non-treated group (Fig. 4).
DISCUSSION
In the previous study, it was reported that human CPP-CARM1 protein was translocated into human MSCs efficiently with biological activity (Jo et al., 2012). In the present study, we confirmed that, mouse CPP-CARM1 protein was well produced and delivered into mESCs efficiently with biological activity. CPPs consisting of short amino acid sequences can deliver biological cargo such as oligonucleotides, small molecules, macromolecules, and proteins (Madani et al., 2011). CPP types and properties are diverse, and the transmissivity of cells differs with the diverse CPP types (Madani et al., 2011). One CPP, oligoarginine, is composed of 5 ‒ 15 arginine sequences, and its cellular uptake potential is very efficient (Mitchell et al., 2000; Tung et al., 2003). The 7R (7x arginine as cationic CPPs)-conjugated CARM1 used in Jo et al. (2012) was connected to a six-histidine tail, whose exogenous proteins were able to be detected by anti-His tag. Using immunocytochemistry of the anti-His tag antibody, we confirmed the introduction of CPP-CARM1 into mESCs (Fig. 2).
In the present study, we found that the CPP-CARM1 protein up-regulated the histone H3R17 methylation in mESCs (Fig. 2). This histone modification activity is known to regulate the pluripotency in mESCs (Wu et al., 2009). As the primary component of chromatin, the histone complex regulates the transcription of pluripotency-related genes and differentiation marker genes through post-translational modification such as acetylation and methylation (Hochedlinger & Plath, 2009; Leeb & Wutz, 2012). CARM1, a member of the PRMT family, is known to up-regulate the transcription level of the Oct4 and Sox2 genes by histone H3 arginine methylation on its genes promoter (Torres-Padilla et al., 2007). Therefore, it can be suggested that CARM1 is a factor that determines cell fate by altering the methylation levels of histone H3 arginine residue 26 when the embryonic blastomeres differentiate into ICM or trophectoderm. Furthermore, when two-cell blastomeres were traced by the injection of Ds-Red tagged CARM1 mRNA, the increased level of CARM1 led to the regulation of the pluripotency-related genes and embryonic polarity (Parfitt & Zernicka-Goetz, 2010). In the present study, we found that pluripotency-related genes were up-regulated in CPP-CARM1 protein-treated mESCs as in previous reports. Based on these reports, it would be considered that the treatment of CPP-CARM1 could change the fate of mESCs into the ICM in reconstituted embryos and might increase the potential for the production of transgenic mice. Okita et al. (2007) reported that the germ line transmission of chimeras had rarely been achieved due to the low expression levels of pluripotency genes in iPSCs. By up-regulating the pluripotency-related gene expression level of iPSCs, it is possible that these types of cells could be more functional pluripotent stem cells for transgenic animals.
We next attempted to elucidate the function of CPP-CARM1 protein in mESCs during in vitro differentiation. Treatment with CPP-CARM1 protein for 1 day increased the expression of pluripotency-related genes, and it may delay the differentiation into EBs. However, when EBs were differentiated for 3 days, the pluripotency-related gene expression levels of the CPP-CARM1-treated and non-treated groups were not different. This result also suggests that the treatment of CPP-CARM1 protein may have a transient effect on the expression of target genes.
In conclusion, we suggest that supplementing CPP-CARM1 protein into culture medium can elevate the expression of pluripotency-related genes in in vitro-cultured mESCs, and it might increase the production efficiency of chimeric mice.