
INTRODUCTION
Alzheimer's disease (AD) is a progressive and the most common neurodegenerative disease. Though the number of AD patients has been increasing recently, there is no fundamental cure. AD is characterized by the impairments of cognitive and memory functions and changes in personality (Selkoe, 1999; Zhang et al., 2011). There are two major pathological features in postmortem brains from patients, which are senile plaques composed of Aβ peptides and neurofibrillary tangles formed of hyperphosphorylated tau (Zhang et al., 2011). Though a small subset of AD cases results from an inherited autosomal dominant gene mutation, most cases of AD are sporadic and age-related. Cellular, molecular, and pathological mechanisms of disease progression and cell death are not fully understood (Reddy, 2009a). However, aging may play a large role in cell death implicated with AD. Oxidative stress increased steadily by aging leads to the accumulation of oxidized proteins, lipids, and nucleic acids (Smith et al., 1996; Gibson & Huang, 2002; Mattson & Liu, 2002; Manczak et al., 2005; Paradies et al., 2010).
Aβ protein plays a critical role in pathogenesis of AD (Zhang et al., 2011). Altered metabolism of Aβ leads to the accumulation and the aggregation of Aβ peptides in the brain. Aβ is derived from APP by sequential cleavage. There are two pathways in APP processing, non-amyloidogenic and amyloidogenic pathway (Gandy, 2005; Zhang et al., 2011). In non-amyloidogenic pathway, APP is processed by α-secretase and γ-secretase. Alternatively, Aβ is derived by cleavage of β-secretase and γ-secretase. Thus, the access of APP to either α- or β-secretase seems to regulate Aβ levels and the pathogenesis of AD (Gandy, 2005; Zhang et al., 2011). Although the specific mechanisms still remain unknown, Aβ promotes neuronal degeneration by increasing the levels of oxidative stress and disturbing cellular energy metabolism (Gibson & Huang, 2002; Mattson & Liu, 2002; Manczak et al., 2005; Querfurth & LaFerla, 2010). Unlike familial AD overexpressing APP by genetic deficits, enhanced amyloidogenic processing of APP may lead to increase oligomeric Aβ in sporadic AD (Smith et al.. 1996; Haass & Selkoe, 2007; Querfurth & LaFerla, 2010).
Since mitochondria is a main source of reactive oxygen species (ROS), its dysfunction can induce complications in ROS processing (Mattson, 2007; Mattson et al., 2008). Increasing number of researches have suggested an association between aging and declining mitochondrial function (Pavlov et al., 2009). Mitochondrial dysfunction has been observed in postmortem brain from AD patients and from AD transgenic mice (Reddy & Beal, 2008). Several studies on mitochondrial enzymes found decreased levels of cytochrome oxidase, pyruvate dehydrogenase and α-ketodehydrogenase in fibroblasts, lymphoblasts and postmortem brains from AD patients (Reddy, 2009b). In addition, free radical production, lipid peroxidation, oxidative DNA damage and oxidative protein damage was increased but ATP production and cell viability was decreased in postmortem AD brains (Parker et al., 1990; Smith et al., 1996; Gibson & Huang, 2002; Maurer et al., 2002).
Mitochondria could be involved in the mechanism by which intracellular Aβ triggers neuronal toxic pathogenesis (Eckert et al., 2003). This idea is supported by the reports, in which Aβ is transported into mitochondria and APP derivatives are accumulated within mitochondria in the brains of AD patients (Fernandez-Vizarra et al., 2004; Lustbader et al., 2004; Devi et al., 2006). Taken together, evidences suggest that mitochondrial dysfunction may play a critical role in the pathogenic mechanisms of Aβ (Eckert et al., 2003; Leuner et al., 2007; Rhein & Eckert, 2007). However, the mechanism of the mitochondrial complex I contributing to Aβ-induced cell death was not fully elucidated.
Here we investigated the role of mitochondria complex I in Aβ toxicity using human SH-SY5Y cells harboring the AD mutant of APP, wild type APP or control vector and found that inhibition of mitochondrial complex I accelerates pathogenic mechanisms of Aβ.
MATERIALS AND METHODS
Human neuroblastoma, SH-SY5Y cells were transfected with DNA constructs expressing human wild type APP (APP-WT) or Swedish APP mutant (APP-swe) using Lipofectamine in serum free media as described (Choi et al., 2004). Human wild type and Swedish mutant (K670N, M671L) APP constructs were the generous gift of Dr. I. Mook-Jung (Seoul National University, Korea). Control cells were transfected with empty vector. SH-SY5Y cells were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 units/mL penicillin and streptomycin at 37℃ in a humidified incubator containing 5% CO2. Culture reagents were purchased from Life Technologies (Grand Island, NY).
Rotenone was dissolved in DMSO (10 mM) and stored in –20℃. Transfected cells were treated with 10-20 μM rotenone diluted in DMEM containing 0.5% fetal bovine serum, 100 units/ml penicillin and streptomycin. Rotenone and other reagents was obtained from Sigma-Aldrich (St. Louis, MO, USA)
SH-SY5Y cells were subcultured in the 24 wells plate. Following stated treatment, cell viability was measured by MTT (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide) reduction assay as described (Choi et al., 1999). MTT reagent was added to the medium 20-24h after drug treatment. After the incubation for 2 h, the equal amount of MTT extraction solution (20% SDS in 50% aqueous dimethylformamide) with media was added and cells were incubated at 37℃ in 5% CO2 for overnight. The absorption of the extract was measured at 570 nm using a microplate reader (Molecular Devices, Sunnyvale, CA).
Cells were washed twice with warm saline buffer (144 mM NaCl, 10 mM HEPES, 2 mM CaCl2, 1 mM MgCl2, 5 mM KCL and 10 mM D-glucose) and incubated with 1 μg/mL dihydroethidium (freshly made from stock solution, 1 mg/mL in DMSO) in saline buffer as described previously (Choi et al., 1999). After 15 minutes of incubation at 37℃ in 5% CO2, cells were washed twice. Cells were detached and moved in 96 well white plate. Cells were quantitated at Ex535 /Em610 (cutoff at 630 nm) using fluorescent microplate reader (Molecular Devices, Sunnyvale, CA).
Cells were washed with PBS, harvested and lysed with lysis buffer containing 62.5 mM Tris-HCl (pH 6.8), 2% w/v SDS, 10% glycerol, 50 mM DTT, 0.01% w/v bromophenol blue, 1 mM PMSF and 5 μg/mL a protinin (Choi et al., 2004). Equal amounts of protein were loaded on 10-15% acrylamide gel and separated by electrophoresis. Samples were transferred onto a PVDF membrane and incubated in 5% skim milk for 1 h. After washing three times with Tris buffered saline with 0.5% Tween 20 (TBST), membranes were incubated with primary antibodies overnight at 4℃. Primary antibodies included rabbit polyclonal antibody against Aβ1-42 (1:3000, Merck, Kenilworth, NJ) and mouse monoclonal antibody against β-actin (Sigma). After washing with TBST, membranes were treated with horseradish peroxidase conjugated secondary antibodies (Merck) for 1 h at room temperature. The bands were specifically detected by enhanced chemiluminescence (ECL, Merck) reaction.
Cells were washed with warm PBS (1M) and incubated with 25 μg/mL propidium iodide in PBS. After 15 minutes, cells were washed twice with PBS. Cells were fixed with 4 % paraformaldehyde /4% sucrose in PBS at room temperature for 15 minutes and washed twice with PBS. Cell were stained with DAPI (1 μg/mL in PBS) and washed. PI and/ or DAPI stained cells were observed using fluorescent microscope (Leica; Heidelberg, Germany). Cells that were stained positive for PI were counted from five representative fields from each well and scored as dead cells. The ratio of PI positive / DAPI positive cells were presented as cell death rate.
RESULTS
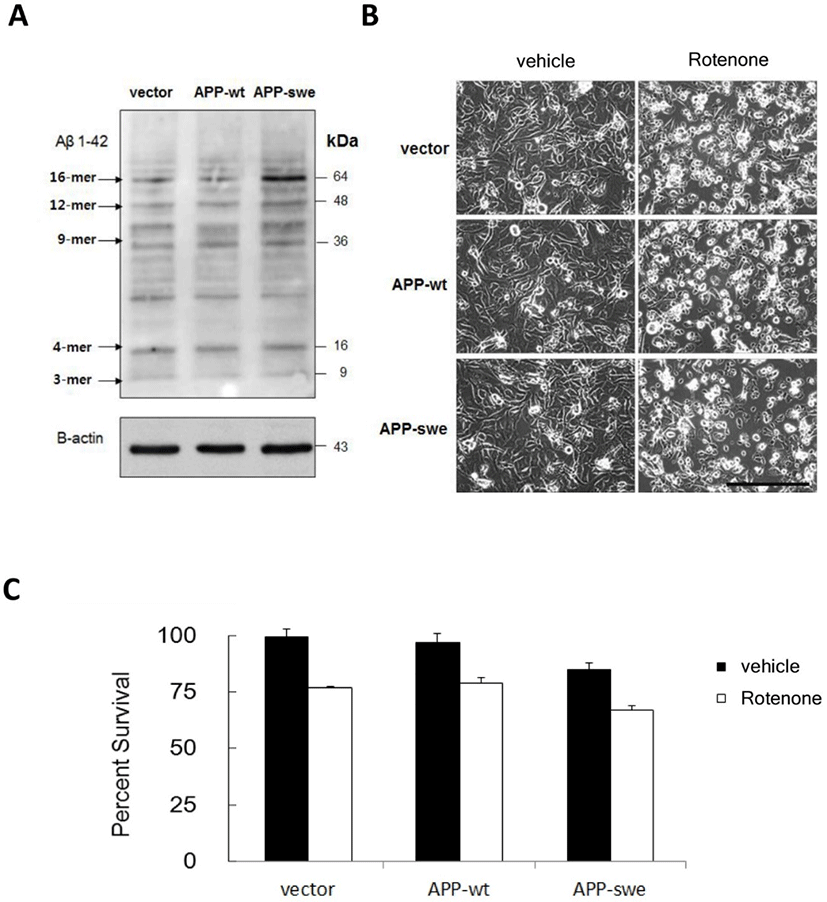
To confirm the generation of Aβ by APP-swe gene transfection, we tested protein levels of Aβ. In SH-SY5Y cells, transfection of APP-swe elevated protein level of Aβ, specially oligomer forms (Fig. 1A). Equal loading of protein was confirmed by β-actin level. The expression of wild type APP did not cause the increase of Aβ. Consequently, cell viability was decreased by the expression of APP-swe, though the difference was not significant and the overall morphology was not altered within 48h of transfection (Fig. 1B, Fig. 1C). We tested the effect of rotenone, complex I inhibitor, to verify the role of mitochondrial inhibition in Aβ toxicity. Rotenone triggered the morphological changes of treated cells. After 18 h treatment with rotenone (20 μM), a number of cells were detached from surface and their processes were fragmented (Fig. 1B, Fig. 1C). For all three groups of transfected cells, rotenone induced loss of cell viability, however there was no significant difference of the effect of rotenone among three groups of cells (Fig. 1B, Fig. 1C).
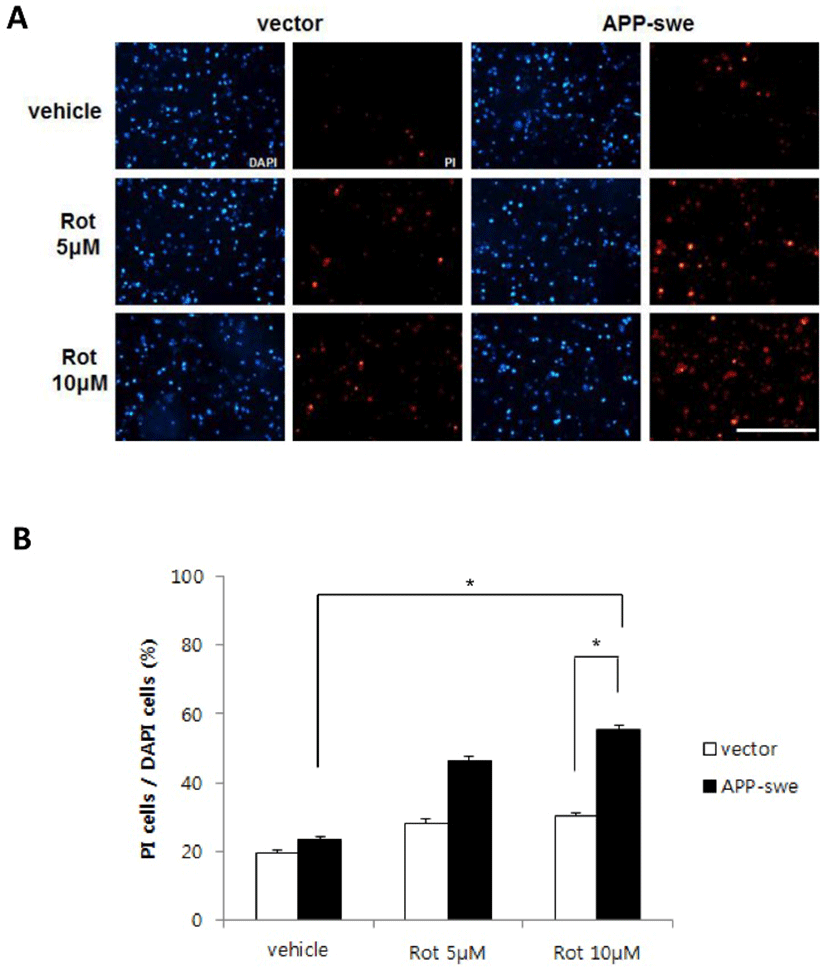
As the difference of the effect of complex I inhibition on cell survival between APP-swe expressing and control cells was not detectable in cell survival assay, we measured cell death rate using propidium iodide (PI) staining. Propidium iodide stains nucleus DNA upon plasma membrane permeabilization which usually occurs in cell death (Lecoeur, 2002). Thus it could be used to identify dead cells. Cells were transfected with either APP-swe or control vector, treated with rotenone 5-10 μM for 18 h, and the numbers of dead (PI-positive) cells per total (DAPI-positive) cells were quantified. The ratio of dead cells was increased by either APP-swe transfection or rotenone treatment (Fig. 2). Moreover, when rotenone was treated in cells expressing APP-swe, cell death was significantly accelerated depending on rotenone dose, compared with vector control, suggesting synergistic effect of mitochondrial inhibition and Aβ toxicity on neuronal cell death (Fig. 2).
Mitochondria is the major hub of cellular energy conversion and major source of ROS generated during the electron transfer process (Gibson & Huang, 2002; Kushnareva et al., 2002; Leuner et al., 2007; Lustbader et al., 2004; Mattson et al., 2008). The majority of ROS derive from complex I and III and complex I dysfunction is found during normal brain aging (Leuner et al., 2007; Petrosillo et al., 2008). Therefore, the decline of complex I activity correlates with ROS level. Previous studies have suggested that generation of ROS and subsequent ROS-mediated signaling may play an essential role in neuronal cell death induced by mitochondrial complex I inhibition (Gibson & Huang, 2002; 7, Choi et al., 1999; Choi et al., 2004; 26-27, Petrosillo et al., 2008). We hypothesized therefore that ROS induced by rotenone contributes to the toxic effect of Aβ in the cell death.
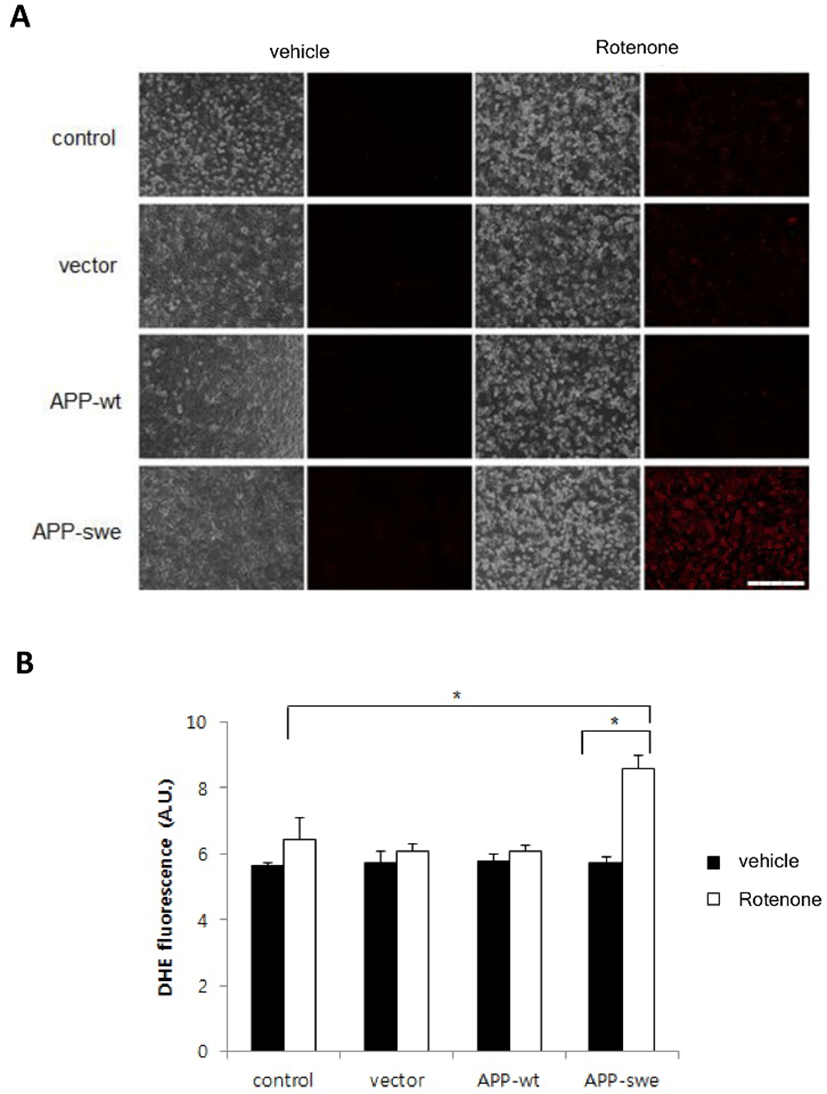
To define whether Aβ increased by overexpressing APP-swe and rotenone treatment affects the level of ROS, cells expressing APP-wt, APP-swe or vector control were incubated with rotenone 10 μM and stained with dihydroethidium (DHE, a cell-permeable superoxide anion indicator). DHE-positive cells were observed using a microscope and superoxide level was measured using microplate reader. As shown in Fig. 3A, expression of APP-wt or APP-swe, by itself, did not increase ROS and rotenone treatment increased ROS level slightly. However, ROS was markedly increased in the cells expressing APP-swe, when they were treated with rotenone (Fig. 3A). Quantitative studies demonstrated that rotenone induces more than 5 times higher increase of ROS in the cells expressing APP-swe than it does in vector control (Fig. 2B).
These results suggest that synergistic effect of mitochondria inhibition and Aβ toxicity on the neuronal cells would be mediated by the elevation of ROS.
DISCUSSION
We intended to investigate if mitochondrial complex I dysfunction can increase the toxicity of Aβ and affect the biochemical mechanism of Aβ toxicity. First, SH-SY5Y neuroblastoma cells were transfected with DNA harboring the human swedish mutant APP gene, the human wild type APP gene or vector control. Aβ oligomer level was increased by APP-swe overexpression in SH-SY5Y cells. Mild treatment of complex I inhibitor, rotenone (5-20 μM), which induces minimum level of cell death, significantly accelerated cell death in the cells transfected with APP-swe. Oxidative stress, demonstrated by oxidation of protein and lipid, is characteristic in AD brain (Varadarajan et al., 2000). Aβ or amyloid plaque binds to metal ions and may mediate generation of ROS. Alternatively, ROS increases Aβ formation in the brain (Leuner et al., 2012). In this study, our data suggest that generation of ROS induced by dysfunction of mitochondria would be accelerated by Aβ and synergistic effect on ROS elevation would be the key factor of AD pathogenesis. This explains why AD is more common in elderly populations, since mitochondrial defect and Aβ are increased during aging.
Interestingly, there was no detectable, rotenone-induced difference in cell viability between APP-swe cells and control cells in MTT assay (Fig. 1), although rotenone-induced cell death was significantly increased in the cells expressing APP-swe compared with control, when tested by PI staining (Fig. 2). MTT assay measures NADH dehydrogenase activity and PI stains cells of which the plasma membrane becomes permeable. Considering distinct mechanisms of these two assay we could explain this controversy. First, a good portion of PI-positive cells may maintain the activity of NADH dehydrogenase even after membrane permeabilization. Second, rotenone and Aβ may accelerate permeabilization of plasma membrane and generation of ROS but not the eventual cell death transiently. This reminds traditional questions on the validity of cell viability tests and the needs to verify cell viability/death with multiple methods.
As expected, complex I inhibition increased ROS in DHE assay (Fig. 3). ROS produced by complex I inhibitor was synergistically increased by Aβ in cells expressing APP- swe. Likewise propidium iodide assay showed dramatic increases of cell death in the cells expressing APP-swe upon treatment of rotenone. This suggests that the combination of Aβ and complex I dysfunction impairs cell viability by increasing oxidative stress. On the basis of these results, we hypothesized that complex I dysfunction associated with increased ROS production potentiates the toxicity of amyloid cascade in AD. A recent study demonstrated ROS increases Aβ formation in the brain, supporting our hypothesis (Leuner et al., 2012). Further study would be needed to elucidate detailed mechanisms involving these combinational effect of mitochondrial defect, Aβ and ROS in AD.
Our results support the idea that the complex I dysfunction increases ROS level and increased ROS level leads to the increase of Aβ level and toxicity. Potentiation of Aβ toxicity may accelerate mitochondria dysfunction, in turn. Based on these results, we suggest complex I dysfunction could be the starting point of the amyloid cascade in sporadic AD, in which the pathogenesis is accelerated by a vicious cycle between complex I dysfunction and Aβ toxicity.