
INTRODUCTION
In reproductive and developmental biology, overcoming infertility remains a critical challenge. The development of egg production and conservation technology is crucial for successfully treating the growing number of patients presenting infertility. However, a considerable number of obstacles persist in the production of numerous functional oocytes. Therefore, to solve some of these challenges, research on in vitro oocyte generation from miniaturized ovaries is critical.
To date, scientists have investigated several strategies based on stem cell research to generate oocytes in vitro (Evans & Kaufman, 1981; Takahashi & Yamanaka, 2006; Takahashi et al., 2007; Hayashi et al., 2011; Hayashi et al., 2012; Aramaki et al., 2013; Nakaki et al., 2013; Kurimoto et al., 2015; Murakami et al., 2016). For example, Hans and colleagues generated oocytes in vitro from mouse embryonic stem cells (mESCs) (Hubner et al., 2003). Hayashi et al. have used a method to aggregate primordial germ cell (PGC)-like cells (PGCLCs) with germline somatic cells to produce oocytes (Hikabe et al., 2016) and Obara and colleagues cultured 12.5 days post coitum (dpc) mouse gonads and produced oocytes (Morohaku et al., 2016). Moreover, oocytes were successfully generated using PGCs and germline somatic cells. The exploitation of an in vitro follicular development system allows the formation of simulated ovaries in the body through co-culture of PGCs and germline somatic cells, thus confirming oocyte formation and embryonic development. In vitro studies regarding ovarian formation techniques have been performed in various species, and research on human oocyte formation remains ongoing. Furthermore, differentiation of human oocytes has been attempted by differentiating induced pluripotent stem cells (iPSCs), which are human pluripotent stem cells, into human PGCs, and by heterogeneous co-culture of established human PGCs (hPGCs) and mouse germline somatic cells (Irie et al., 2015; Sasaki et al., 2015). Although a few hundred follicles were obtained in a single ovary in vitro, the inner follicles did not receive adequate signals and were unable to fully develop, as distinct blood vessels were not developed like those seen in vivo. Thus, isolation of single follicles is necessary to stimulate follicle growth and maturation. These problems can be solved by establishing a miniaturized ovary capable of producing fewer follicles. By forming a miniaturized ovary, signals can reach the inner part, allowing nutritional supply. In addition, challenges in isolating individual follicles can be overcome, allowing a smooth transition to the in vitro maturation (IVM) process. Eventually, it will be easier to observe ovarian formation, and large amounts of oocytes can be efficiently obtained without issues such as oocyte loss.
Herein, we established a culture system producing a miniaturized ovary. First, we optimized the culture conditions for follicle formation from mouse gonads and then optimized the system for dissociation of gonads by employing suitable enzymes to generate miniaturized ovaries containing 5–30 follicles. In vitro oocytes from miniaturized ovaries could mature into MII oocytes.
MATERIALS AND METHODS
Pregnant female ICR mice at 12.5, 13.5, and 14.5 dpc were employed to isolate gonad for dissociation experiments. The following reagents and chemicals were procured from respective suppliers: Dulbecco’s modified Eagle’s medium (1×) (DMEM; 11965-092, Gibco, Grand Island, NY, USA), phosphate-buffered saline (PBS; Gibco, cat. no. 10010-023), Minimum Essential Medium Eagle, alpha modification (1×) (α-MEM; cat. no. 12571-063 and 32571-036, Gibco), 2-mercaptoethanol (55 mM; cat. no. 21985-023, Gibco), fetal bovine serum (FBS; cat. no. 16000-044, Gibco), Pen Strep Glutamine (100×) (cat. no. 10378-016, Gibco), ascorbic acid (150 mM; cat. no. G0394, TCI), ICI182 and 780 (cat. no. 1047, Tocris), follicle-stimulating hormone (FSH; cat. no. F4021, Sigma-Aldrich, St. Louis, MO, USA), epidermal growth factor (EGF; cat. no. 2028-EG-200, R&D Systems, Minneapolis, MN, USA), polyvinylpyrrolidone (PVP; cat. no. PVP360100G , Sigma-Aldrich), human chorionic gonadotropin (1,200 IU mL–1) (hCG; cat. no. C8554, Sigma-Aldrich), collagenase type IV (1 g mL–1; cat. no. 17104-019, Gibco), trypsin-EDTA, 0.25% (cat. no. 25200-072, Gibco), StemPro-34 SFM (1×), liquid (cat. no. 10639-011, Gibco), and hyaluronidase (cat. no. D8418, TCI).
The following list of equipment was employed to perform the experiments: Clean bench (cat. no. JSCB-1500SB, JSR), water bath (cat. no. JSWB-22T, JSR, Gongju, Korea), CO2 incubator ( JP/SMA-165D , Astec, Seoul, Korea), stereoscopic microscope (cat. no. CKX53, Olympus, Tokyo, Japan), a suction machine (cat. no. TFS-30, Unim, Gunpo, Korea), centrifuge (cat. no. KLG122618050210, Labogene, Allerød, Denmark), a hemocytometer (cat. no. 2960408,Paul Marienfeld, Lauda-Königshofen, Germany), micropipettes (cat. nos. EP3120000011, EP3120000020, EP3120000038, EP3120000046, EP3120000054, EP3120000062, Eppendorf, Hamburg, Germany), sterile plastic tips (10 μL, 200 μL, 1,000 μL; Thermo Fisher Scientific, Waltham, MA, USA), pipettors, sterile plastic pipettes (5, 10, 25, and 50 mL), 15-mL conical SW Jang, HW Choi centrifuge tubes (cat. no. 338652 or equivalent, Thermo Fisher Scientific), 50-mL conical centrifuge tubes (cat. no. 338652 or equivalent, Thermo Fisher Scientific), four-well culture plates (cat. no. 176740, Thermo Fisher Scientific), Petri dish 35 mm (cat. no. 353001, BD-Falcon), Petri dish 100 mm (cat. no. 353003, BD-Falcon), 24 well-tissue culture plates (cat. no. 3526, Costar, NY, USA), Transwell-COL membrane inserts (cat. no. 3496, Costar), sterile syringes (0.5 mL; cat. no. 328821, BD, New Jersey, USA), tungsten wire (cat. no. W-461387, Nilaco, Tokyo, Japan), cell strainer (40, 70, 100, 150 μm ; cat. nos. 93070, 93100, SPL, Pocheon, Korea), and 35 mm hexagon U-bottom plate (cat. no. H2951000, StemFIT 3D, Hanam, Korea).
No statistical methods were used to predetermine sample size. The experiments were not randomized. The investigators were not blinded to allocation during experiments and outcome assessment.
All methods were performed in accordance with relevant guidelines and regulations of Jeonbuk National University. All experimental protocols were approved by the Institutional Animal Care and Use Committee of Jeonbuk National University (approval numbers CBNU-2019-00395). Pregnant female outbred albino ICR mice were purchased from Company of Samtako (Osan, Korea). Pregnant females were killed by cervical dislocation to obtain E12.5, E13.5 and E14.5 embryos. Only from female embryos, the gonads were isolated and then dissociated for generation of miniaturized ovaries. This study was carried out in compliance with the ARRIVE guidelines (http://www.nc3rs.org.uk/page.asp?id=1357).
Female fetal gonads without mesonephros were cultured in Transwell-COL membranes (3.0-μm pore size, 24-mm diameter, Corning, Corning, NY, USA) soaked in α-MEM-based IVDi medium (α-MEM supplemented with 2% FBS, 150 μM ascorbic acid, 1× penicillin/streptomycin/ L-glutamine, and 55 μM 2-mercaptoethanol). From days 5 to 11 of culture, 10 μM ICI182780 was added to the α-MEM-based IVDi medium (henceforth, referred to as α-MEM). On day 4 of culture, the culture medium was changed to StemPro-based IVDi medium (StemPro-34 SFM supplemented with 10% FBS, 150 μM ascorbic acid, 1× penicillin/streptomycin/L-glutamine, and 55 μM 2-mercaptoethanol). From days 7 to 10 of culture, 500 nM ICI182780 was added to the Stempro-34-based IVDi medium (henceforth, referred to as StemPro). On day 21 of culture, individual secondary follicles (2FLs) were manually dissociated using sharpened tungsten needles. Gonads were cultured for 21 days at 37℃ under 5% CO2 and 95% air. Approximately half of the medium in each well was replaced with fresh medium every other day.
Electrically sharpened tungsten needles were used to isolate the individual 2FLs. The interstitial cells between 2FLs were carefully removed. Then, 2FLs were separated from the ovaries and placed at regular intervals.
Single 2FLs on Transwell-COL membranes were soaked in IVG-α-MEM medium (α-MEM supplemented with 5% FBS, 2% polyvinylpyrrolidone, 150 μM ascorbic acid, 1× penicillin/ streptomycin/L-glutamine, 100 μM 2-mercaptoethanol, 55 μg mL–1 sodium pyruvate, 0.1 IU mL–1 FSH, 15 ng mL–1 BMP15, and 15 ng mL–1 GDF9). After 2 days of culture, the medium containing BMP15 and GDF9 was withdrawn, and follicles were then incubated in 0.1% type IV collagenase. After washing several times with α-MEM supplemented with 5% FBS, the follicles were cultured in IVG-α-MEM without BMP15 and GDF9. On day 11 of culture, cumulus-oocyte complexes grown on the membrane were picked using a fine glass capillary, cultured on Transwell-COL or Millicell membranes for another 9–13 days at 37℃ in medium, under 5% CO2 and 95% air. Then, 1 mL of medium was added to the inside and outside of membrane inserts. Approximately half of the medium in each well was replaced with fresh medium every other day.
Cumulus-oocyte complexes were transferred to IVM medium (α-MEM supplemented with 5% FBS, 25 μg mL–1 sodium pyruvate, 1× penicillin/streptomycin, 0.1 IU mL–1 FSH, 4 ng mL–1 EGF, and 1.2 IU mL–1 hCG ). After 16 h of culture, swollen cumulus cells were stripped from oocytes by treatment with hyaluronidase, and then MII oocytes were determined by first polar body extrusion.
In brief, oocytes were fixed for 4 h at 20℃ in 4% paraformaldehyde. The cells were then washed with PBS and treated with PBS containing 10% normal goat serum and 0.03% Triton X-100 for 45 min at room temperature. The primary antibodies used were anti-Ddx4 (polyclonal, 1:500; Abcam, Cambridge, MA, USA), anti-Oct3/4 (monoclonal, 1:500; Santa Cruz Biotechnology, Dallas, TX, USA), and GDF9 (polyclonal, 1:500; Invitrogen, Carlsbad, CA, USA). For detection of primary antibodies, fluorescently labeled (Alexa Fluor 488 or 568; Molecular Probes, Eugene, OR, USA) secondary antibodies were used according to the manufacturer’s specifications. All immunofluorescence samples were analyzed using a confocal microscope (LSM700, Carl Zeiss, Oberkochen, Germany).
RESULTS
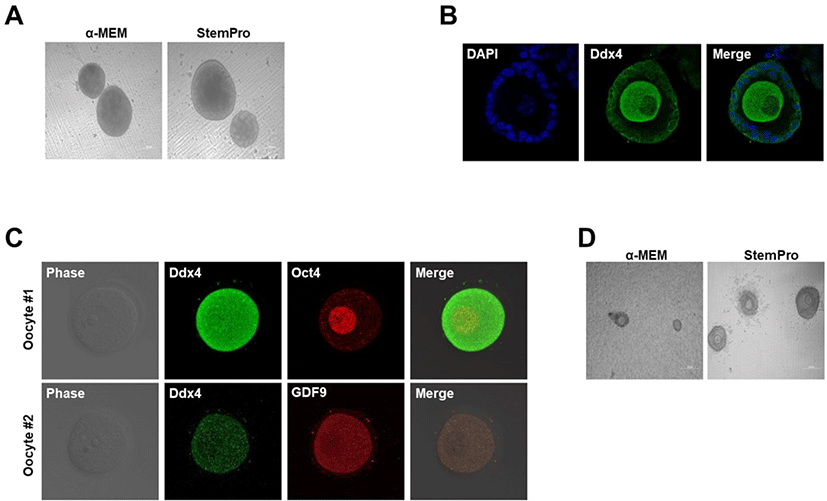
In our early experiments, female gonads obtained from E12.5–E14.5 mice were cultured in α-MEM or StemPro medium for 5 weeks on Transwell-COL membranes. First, we assessed the efficiency of follicle formation in the two culture media. Two control groups were prepared: one cultured using the α-MEM-based culture medium (Morohaku et al., 2016) and the other cultured in StemPro medium (Hikabe et al., 2016). On employing the two cultures for preserving gonads, the formation of follicles was assessed, and secondary follicle formation was observed (Fig. 1A and B). Follicles generated using two different media gradually matured and produced oocytes (Fig. 1B). The in vitro oocytes expressed Oct4 and Ddx4, as well as the oocyte-specific marker GDF9 (Fig. 1C). Oct4 is expressed in the oocyte core, whereas Ddx4 is expressed on the oocyte (Noce et al., 2001; Saitou et al., 2002; Tanaka et al., 2004; Okamura et al., 2008; Pisarska et al., 2011; Sabour et al., 2011). These data revealed that, in both media, the cultures could form normal functional oocytes. Collectively, the gonads from the period between 12.5, 13.5, and 14.5 dpc could develop into follicles in vitro on Transwell-COL membranes, irrespective of the medium employed. The follicle formation efficiency did not significantly differ between the two types of culture media. However, in vitro ovaries in StemPro medium contained considerably more follicles, demonstrating more rounded granulosa cells in the IVG medium than those in the α-MEM medium. Additionally, we confirmed that the granulosa cells were further expanded during the IVG process using StemPro-based medium (Fig. 1D). Therefore, we used StemPro medium to perform all further experiments.
Next, to determine whether a single follicle and oocyte possessing a three-dimensional (3D) structure could be formed, we cultured single cells dissociated from mice gonads at 12.5, 13.5, and 14.5 dpc on a Transwell-COL dish using StemPro medium. Additionally, to confirm enzyme efficiency for gonad dissociation into single cells, two enzymes were assessed, i.e., trypsin-EDTA and collagenase IV. First, we used trypsin-EDTA to dissociate the gonads into single cells, which demonstrated high dissociation efficiency into single cells. We cultured single cells on Transwell-COL after trypsin-EDTA treatment to confirm dissociation of mice gonads at 12.5, 13.5, and 14.5 dpc. Spherical cells, which differed from the germline somatic cell shape, were found to grow on Transwell-COL membranes. We analyzed cultured globular cells, and expression levels of the germline cell marker Ddx4 were confirmed by immunocytochemistry (Fig. 2A). Spherical cells displayed Ddx4 expression on the cell surface; this was demonstrated by 12.5 dpc gonads, as well as 13.5 and 14.5 dpc gonads (Fig. 2A). Reportedly, gonadal PGCs are known to demonstrate oogonia, which can differentiate into meiotic cells through multiple cell divisions during migration to the genital ridge (Hayashi & Saitou, 2013; Saitou & Miyauchi, 2016). These results demonstrated that, when single cells dissociated from gonads and PGCs and gonadal somatic cells were co-cultured in Transwell-COL, co-cultured single cells could differentiate into oogonia-like cells expressing Ddx4, without follicle formation (Lei et al., 2006).
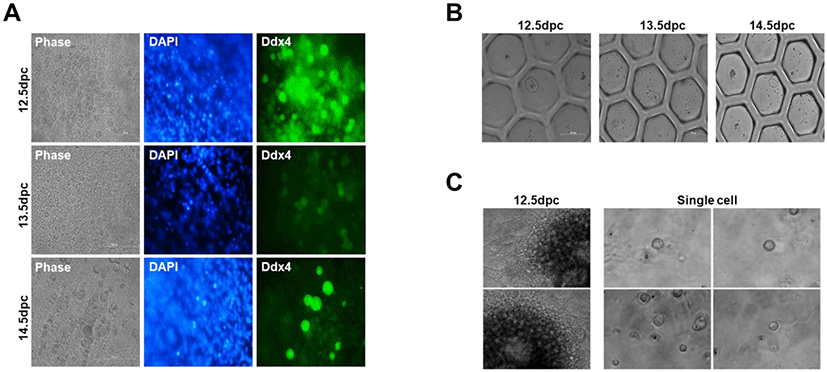
Then, we aggregated and cultured PGCs and germline somatic cells on a U-bottom plate, as we failed to generate a 3D structure on Transwell-COL membranes. Single cells from gonads were seeded at a density of 2.5 × 105 per well in a hexagon-shaped U-bottom plate. The cells aggregated after 1 day, but no follicle formation was observed (Fig. 2B); these results were similarly observed for 12.5, 13.5, and 14.5 dpc gonads. As another method for 3D culturing, we used Matrigel, which is widely used for organoid culture or 3D tissue culture (Lang et al., 2001; Pierzchalska et al., 2012; Dolega et al., 2015). Similar to previous results, it did not facilitate ovary formation (Fig. 2C). These results indicated that once the gonad is dissociated into a single cell, ovary formation is impossible even if clustered again.
We employed several strategies to generate a minimally-sized ovary that can form a single follicle. Accordingly, the gonad was manually separated into three parts, and all three parts formed the ovary, producing fewer follicles than the control (Fig. 3A). In addition, follicles formed in this manner generated superior oocytes. Furthermore, the oocytes within the ovary were well-developed, similar to the oocytes on the outside. Signals and nutrients were well transmitted to the inner part of the ovary. However, the method failed to generate fewer ovarian follicles. Therefore, loss of follicles can still occur during the isolation process. During this method, we used a thin syringe to mechanically cut the gonad into the smallest fragments possible. We then assessed ovary formation and observed that follicles were produced when gonads were cut to the minimum size without isolation into a single cell state. Accordingly, all gonads adequately formed ovaries; however, numerous follicles were generated. Moreover, the number of follicles appeared to random in the in vitro ovaries obtained (Fig. 3B). This method was unsuitable for generating uniform follicles in all ovaries, and it was difficult to individually isolate follicles. Based on these findings, it was confirmed that ovaries were formed even when the gonad was separated into a gonadal mass without isolation into a single cell. Next, we attempted to confirm whether ovary formation occurred when smaller gonadal masses were achieved using a collagenase IV suspension rather than mechanical separation. Using the collagenase IV enzyme, we observed that gonads were dissociated into an appropriate gonad mass that could form ovaries. In the present study, the efficiency of dissociation into an appropriate gonadal mass was increased by controlling the collagenase IV treatment time and suspension. Following collagenase IV treatment for more than 2 min, gonadal masses were broken into single cells; when treatment was performed for less than 2 min, ovaries producing numerous follicles were formed. After approximately 2 min of collagenase IV treatment in suspension, a few minimally crushed masses could be identified. We cultured the masses and found that ovaries with a fewer number of follicles were formed. The ovaries generated using this method were smaller in size than those generated using the previous methods; however, a random number of follicles were produced (Fig. 3C). This strategy successfully reduced ovary size and follicle number; however, precise standardization remained challenging.
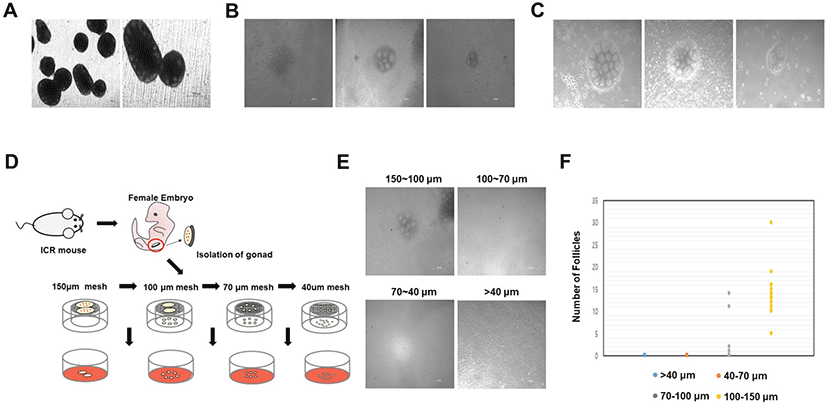
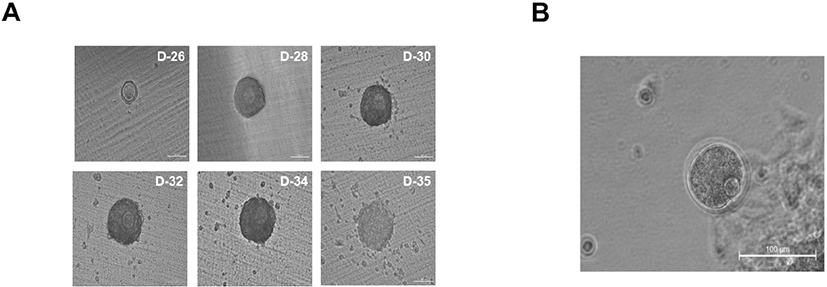
Finally, we cultured gonad tissues after breaking them down to a uniform size using 150, 100, 70, and 40 μm meshes. Collagenase IV treatment was performed for 2 min, followed by repeated suspension two or three times; in the absence of adequate collagenase treatment, all gonads will be similar in size as control gonads. In order to determine the minimum mass size that can accurately form follicles, the gonad was cultured by dividing it into four groups after passing through 150, 100, 70, and 40 μm meshes. Thus, gonad masses were cultured in four groups of 150–100, 100–70, 70–40, and 40 μm or less (Fig. 3D). When cultured by breaking the gonad using a 40 μm mesh, the gonadal mass formed ovaries but no follicles (Fig. 3E). Additionally, the gonadal mass passed through the 70 μm mesh failed to generate follicles (Fig. 3E). In contrast, the gonadal mass passed through the 100 μm mesh and retained in the 70 μm mesh only formed four ovaries, which produced follicles (Fig. 3F). However, owing to the low ovarian production rate, it was challenging to produce numerous oocytes. Gonadal mass passed through the 150 μm mesh and retained in the 100 μm mesh was equal or slightly larger than 100 μm sized mass, formed miniaturized ovaries, and contained an average of 12 uniform follicles per ovary (Fig. 3F). In gonadal masses ranging between 150 μm and 100 μm in size, ovaries generated normally developed follicles during IVG (Fig. 4A). These results indicate that a minimum gonadal mass of 100 μm is required to form an ovary capable of generating follicles. In other words, when the gonad is crushed to a size less than 100 μm, the cells are damaged, and ovaries cannot be formed. In this experiment, we used collagenase IV treatment, manual cutting, and passage through a mesh. On using a mesh, we were able to identify the most efficient and suitable size. Undoubtedly, the method was less time-consuming, resulted in reduced cell loss during isolation, and produced ovaries with fewer follicles. Although there are technical limitations to generating ovaries that form single follicles, these issues can be surpassed. Ten days after IVG, oocytes were well-developed into germinal vesicle (GV) oocytes (Fig. 4A). GV oocytes were treated with IVM medium which used to resume meiosis. After 16 h, the oocytes extruded the first polar body (Fig. 4B).
DISCUSSION
The generation of a large number of in vitro oocytes is a critical step in overcoming infertility. Therefore, developing the most efficient and appropriate method to produce numerous in vitro oocytes remains crucial in reproductive biology and developmental biology. To date, the generated ovaries have produced hundreds of follicles, such that the inner follicles fail to generate normal oocytes owing to a lack of signal, nutrient factors, and lack of space for cell growth (Hikabe et al., 2016; Morohaku et al., 2016). Accordingly, it is important to isolate several follicles generated in the ovary. Culturing several ovaries that produce fewer follicles in vitro would be less time-consuming and less complex, increasing the production of GV oocytes. Moreover, it will be easier to observe ovary formation in vitro. Thus, it will be possible to overcome the infertility problem by efficiently obtaining numerous oocytes.
In the present study, Hayashi and colleagues showed that more follicles were generated in in vitro ovaries when the aggregates with PGCs and somatic cells were cultured in StemPro medium (Hikabe et al., 2016). We compared α-MEM and StemPro to determine which media would be ideal. There was no noticeable difference between cultures using the two media; however, StemPro appeared to generate a small number of follicles with more rounded oocytes. The gonads cultured under these conditions formed a normal ovary, well-produced oocytes. These oocytes expressed Oct4 (pluripotent marker), Ddx4 (germ cell marker), and GDF9 (oocyte-specific marker). Notably, both cultures must be initiated with α-MEM media. The gonads are composed of PGCs and somatic cells. We first devised a method to break the gonads into single cells on a Transwell-COL membrane. We employed 0.25% trypsin-EDTA, which produced a more significant number of intact integrins and faster dissemination (Brown et al., 2007). Single cells that were completely broken following trypsin treatment failed to form ovaries, generating only oogonia-like cells. Next, when single cells were aggregated on the U-bottom plate, we assessed whether they could form normal ovaries. Hayashi and colleagues isolated PGCs into single cells and then aggregated them with germline somatic cells to successfully form a simulated ovary (Hikabe et al., 2016). They used PGCs, differentiated from ESCs, and these cells appeared to be similar to the PGC stage of E 9.5 in vivo. In contrast, PGCs used in this study were isolated from the germline 12 days after implantation. Additional investigations are needed to elucidate the developmental process of PGCs and the effect of single-cell dissociation. Single cells from the 12.5, 13.5, and 14.5 dpc gonads could be aggregated into Transwell-COL or U-bottom plates; however, follicle formation could not be achieved. These cells formed spheres rather than ovaries. Spherical cells were examined for Ddx4 expression. Based on immunochemistry, surface Ddx4 expression was confirmed. The ovary was not formed in the single cell state, with differentiation into oogonia-like cells observed (Hickford et al., 2011; Saitou & Miyauchi, 2016). In several species, germ cells are connected by cytoplasmic bridges and can share cytoplasmic contents between germ cells at different stages of development until a mature gamete is formed. All germ nuclei share a common cytoplasm— this is not an ideal condition for effective transcriptional control (Fawcett, 1961; Gondos, 1973; Gondos & Conner, 1973; Pepling et al., 1999; Ventelä, 2006; Pushpa et al., 2017). Pepling and Spradling analysis in mouse ovaries showed the presence of intercellular bridges (ring canals) between the ovarian germ cells at E11.5 and E17.5 (Pepling et al., 1999).
In conclusion, we observed that PGCs with somatic cells could differentiate into oogonia-like cells when dissociated into single cells using gonadal somatic cells. Furthermore, we revealed that when gonads were dissociated into single cells, ovary formation could not be achieved following aggregation of PGC and gonad somatic cells in the U-bottom plate.
Several methods have been used to form ovaries with a small number of oocytes. We attempted to manually cut the gonadal tissues into three equal parts. In this method, the number of oocytes from the ovaries was lower than that of the control. However, prior to the IVG process, it appears that signals are not well transmitted to the inner part of the ovary; hence, inner oocytes fail to develop normally. Next, we manually cut the gonads as feasible using a syringe. This method generated ovaries that formed fewer follicles, which was undoubtedly more effective than the previous method. However, there were difficulties in achieving a uniform number of follicles, resulting in a considerable number of follicles. We then employed collagenase type IV, frequently used for tissue dissociation. Following treatment with collagenase IV, the cells broke down into an appropriate gonadal mass. We dissociated the gonadal mass considering only the collagenase IV treatment time and suspension. The number of oocytes formed per in vitro ovary was random, and the size tended to vary. However, the ovaries formed few follicles. Hence, we developed a more efficient method to evenly generate a small number of follicles. Previously, Kim and colleagues generated ovaries using a mesh to isolate follicles from ovarian tissues; they optimized the system for follicle isolation from ovarian tissue using diameters of 0.254 mm and 380 μm meshes (Kim et al., 2018). We attempted to generate miniaturized ovaries in vitro, and dissociated gonads were sequentially passed through 150, 100, 70, and 40 μm meshes followed by seeding on Transwell-COL membranes. Accordingly, we observed that ovaries were formed only with the 150–100 μm and 100–70 μm masses, with no ovaries observed in groups below 70 μm. In the 100–70 μm group, except for 4 ovaries, most did not form ovaries. Thus, the probability of reproduction was low. In the 150–100 μm group, approximately 12 follicles were formed on average, and the follicles were well-developed following even the distribution of appropriate signals and nutrients.
Our in vitro system will be useful for developing an ovary model to visualize and overcome cell loss and time consumption encountered during the isolation process. Therefore, our system can generate more well-developed oocytes for fertilization. This method would help solve infertility challenges for all potential parents and will significantly impact national fertility.