INTRODUCTION
Chondrocytes are the cellular component of cartilage, responsible for generating and maintaining its extracellular environment (Muir, 1995). Cartilage has multiple functions such as providing cushion on the articular surfaces of joints, providing a template for the formation of endochondral bone, and contributing to fracture repair (Sandell & Aigner, 2001; Karsenty & Wagner, 2002). However, cartilage defects have only a limited intrinsic healing capacity (Hellingman et al., 2011). Thus, the cell-based regeneration of (osteo) chondral defects presents a major challenge (Pelttari et al., 2008; Fong et al. 2010; Huang et al., 2010).
Human mesenchymal stem cells (MSCs) with multiple differentiation potentials are a promising alternative cell source for cartilage regeneration (Csaki et al., 2008; Chen et al., 2009). The differentiation of mesenchymal cells into chondrocytes takes place along a multistep pathway (Shum & Nuckolls, 2002). The steps in this pathway include the recruitment of mesenchymal chondroprogenitor cells, the subsequent condensation of these mesenchymal cells, followed by the frank differentiation of the condensed mesenchymal cells into chondrocytes. Cells within these condensations commit to the chondrogenic lineage, acquire spherical cell morphology and induce expression of the essential chondrogenic transcription factor Sox9. Sox 5 and Sox6 cooperate with Sox9 to control chondrogenesis and are themselves under the transcriptional control of Sox9 (de Crombrugghe et al., 2000; Kawakami et al., 2006). Together, these transcription factors activate transcription of the major chondrogenic matrix genes, collagen type II (COL II) and aggrecan. Furthermore, increased glycosaminoglycan (GAG) content is another marker of the chondrogenic extracellular matrix (ECM) (Bobick et al., 2009; Chen et al., 2009). In the growth plate of skeletal elements that undergo endochondral ossification, several layers of chondrocytes then become flattened and the cells proliferate mainly unidirectionally (Shum et al., 2002; Woods et al., 2007). These cells then stop proliferating, change their genetic program and become hypertropic (Pelttari et al., 2008). The ECM of the most advanced hypertrophic chondrocytes becomes mineralized before these cells undergo apoptosis and are replaced by the cells that will become the constituent cells of bones (Fong et al., 2010).
Chondrogenic induction in vitro stands as a special culture system achieved by forcing aggregation for mesenchymal cells or chondroprogenitor cells to generate a ‘micromass’ or ‘pellet’ culture and treating this with transforming growth factor-β (TGF-β) superfamily members (Boeuf & Richter, 2010; Vater et al., 2011). TGF-β promotes cartilage-specific gene expression through intracellular signaling cascades involving SMAD proteins, and the mitogen activated protein (MAP) kinases (Augello & De Bari, 2010; Li et al., 2010; Arita et al., 2011; Hellingman et al., 2011). The therapeutic potential of MSCs for cartilage repair is clear (Csaki et al., 2008; Pelttari et al., 2008; Chen et al., 2009). However, the requirements and conditions for effective induction of chondrogenesis in MSCs and for the production of a stable cartilaginous tissue by these cells are far from being understood. Thus, gaining a better understanding of signaling pathways that regulate these conditions is essential.
A C2 domain is a protein structure domain involved in targeting protein to cell membrane. The C2 domain is found in many cellular proteins involved in signal transduction or membrane trafficking. C2 domains are unique among membrane targeting domains in that they show wide range of lipid selectivity for the major components of cell membranes, including phosphatidylserine and phosphatidylcholine (DiNitto et al., 2003). This C2 domain is about 116 amino-acid residues and is located between the two copies of the C1 domain in Protein Kinase C (PKC) and the protein kinase catalytic domain. Regions with significant homology to the C2 domain have been found in many proteins (Corbalán-García & Gómez-Fernández, 2010).
Although the function of C2 domain in chondrogenesis is unknown, C2 domain may play a role in signaling pathways that regulate chondrocyte differentiation. The present study was undertaken to reveal whether the C2 domain is involved in signaling processes of chondrogenesis.
MATERIALS AND METHODS
Human MSCs were purchased from Lonza (Walkersville, MD). The cells were expanded in low-glucose DMEM containing 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml) in a 5% CO2 incubator at 37°C. Normal human fibroblast (NHFB) were obtained from Chungnam National University and cultured in DMEM containing 10% fetal bovine serum, penicillin (100 units/ml), and streptomycin (100 μg/ml). All culture media and supplements were obtained from Gibco (Carlsbad, CA).
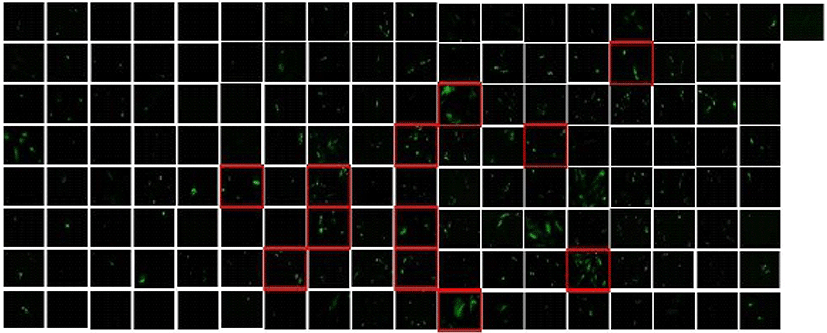
The C2 domain library containing 145 kinds was manufactured using the gateway adenovirus system (Nochi et al., 2004; Park et al., 2007). That adenovirus library was then infected to hMSCs individually. Final candidates were classified and selected by the degree of their effects on morphological changes.
Chondrogenic differentiation of the hMSCs was initiated in a micromass culture system (Zhang et al., 2010; Vater et al., 2011). Cells were dissociated for single-cell suspension stating at density of 2.0×107 cells/ml, and a 10-μl drop of this cell suspension was placed in the center of a culture dish. The cells were allowed to adhere at 37°C for 2 h, followed by the addition of chondrogenic medium (high-glucose DMEM containing 100 nM dexamethasone (Sigma, St. Louis, MO), 50 μg/ml ascorbic acid-2-phosphate (Sigma), 1% penicillin streptomycin, and ITS-Premix (6.25 μg/ml insulin, 6.25 μg/ml transferrin, 6.25 μg/ml selenous acid, 5.33 μg/ml linoleic acid and 1.25 mg/ml bivine serum albumin; BD Biosciences, Bedford, MA) with or without 10 ng/ml TGF-β3 (R&D systems, Minneapolis, MN). After 24 h, the cell droplets coalesced and became spherical. The medium was changed every 2 days.
Human MSCs at 80% confluence were transfected with 100 MOI of Adv-GFP or Adv- PKCη-C2 domain for 2 h, and media were changed with fresh DMEM + 10% FBS. Seven day after infection, cell morphology was examined using an inverted fluorescence microscope (IX81, Olympus, Japan) coupled to a CCD camera (Olympus DP71). For micromass culture, transfected cells were seeded 24 h after infection.
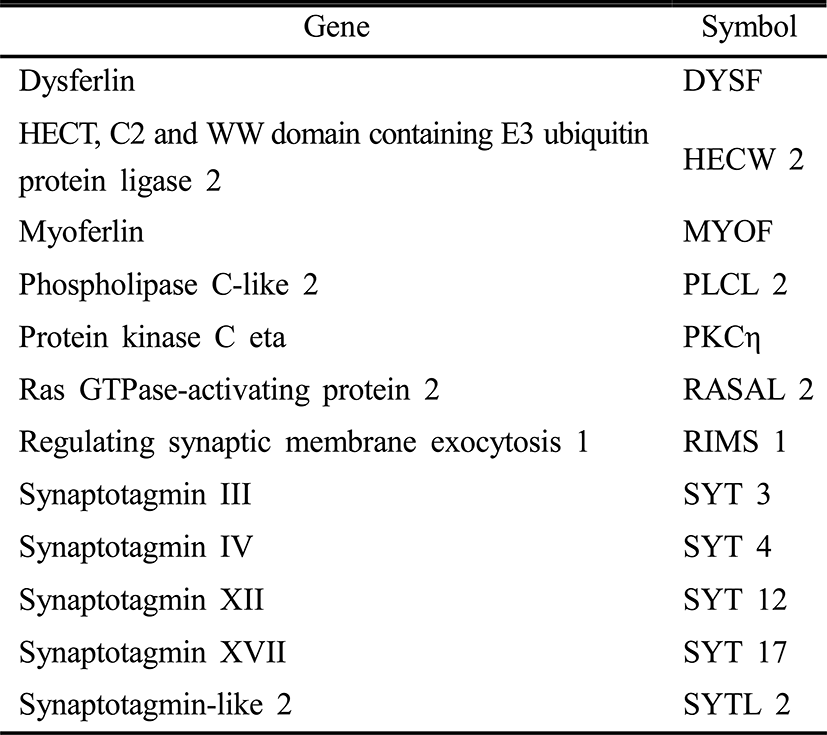
Total RNA was isolated with the TRIzol reagent (Invitrogen, Carlsbad, CA) and 1 μg RNA was reverse transcribed for synthesis of first cDNA strand. Synthesized cDNA was used as a template in PCR using Taq polymerase (Takara, Japan) with gene specific primers (Table 2). PCR products were separated on 1.5 % agarose gel and visualized by ethidium bromide staining.

Cells were lysed in lysis buffer containing 50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 1% NP-40, 0.02% sodium azide, 0.5% sodium deoxycholate, 0.1% SDS, 100 μg/ml phenylmethylsulphonyl fluoride, 0.5 μg/ml leupeptin, 1 μg/ml aprotinin. After a brief sonication, the lysates were clarified by centrifugation at 12,000×g for 30 min at 4°C. Protein concentrations were determined with a BCA protein assay kit (Pierce Biotechnology, Inc., Rockford, IL). Equal amounts of protein were loaded on 10% SDS-polyacrylamide gel and transferred to a nitrocellulose membrane. Membranes were blocked with 5% skim milk in TBS containing 0.1% Tween-20 for 2 h at room temperature and incubated with the appropriate primary and secondary antibodies. Labeled proteins were detected using ECL Western blotting detection reagents (Amersham Biosciences, Piscataway, NJ). Antibodies used were: COL1A1, COL2A1, and PKCη (Santa Cruz Biotechnology, Santa Cruz, CA), α-tubulin (Sigma).
Micromasses were fixed in 4% paraformaldehyde for 3 h, then dehydrated with ethanol, cleared with xylene and embedded in paraffin. Sections at 5 μm were cut and mounted on glass slides. Sections were deparaffinized and hydrated to distilled water, followed by staining with hematoxylin and eosin or Alcian blue and fast red. Finally sections were brought to xylene solution with several dehydration steps and mounted.
Chondrogenic differentiation was measured by Alcian blue staining of sulfated cartilage glycosaminoglycans. Cells were fixed in 4% paraformaldehyde for 10 min and then incubated with 0.1% HCl-Alcian blue for 1 h. Excess stain was washed off with distilled water, and pictures were taken. To evaluate staining intensity, Alcian-blue-bound sulfated glycosaminoglycans were extracted with 6 M guanidine-HCl, and quantified by measuring the absorbance of the extracts at 620 nm.
Binding of peanut agglutinin (PNA) was used as a specific marker for precartilage condensation. Cells were fixed in 4% paraformaldehyde for 10 min and then incubated with 100 μg/ml FITC conjugated PNA (Sigma) for 1 h. PNA binding was visualized using FITC fluorescence.
RESULTS
To screen whether C2 domain can differentiate hMSCs to chondrocyte, 145 C2-domain containing adenovirus were infected to hMSCs individually. Seven days after transfection, primary candidates were selected by the degree of their effects on morphological changes (Fig. 1). As shown in Table 1, 12 C2 domains were selected for further study.
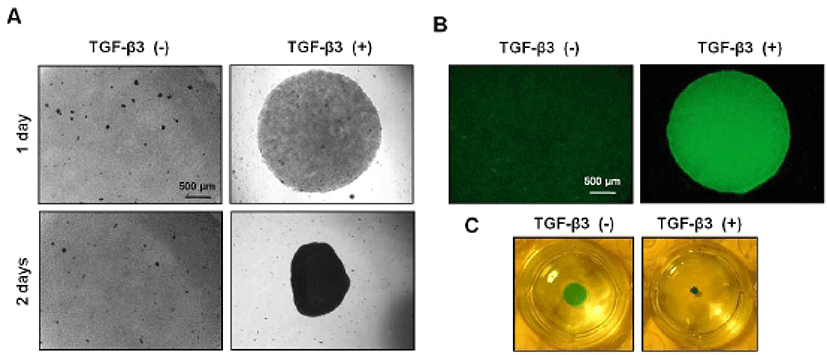
To induce chondrogenesis of hMSCs, micromasses were cultured in chondrogenic medium and stimulated with TGF-β3 (10 ng/ml). In most MSC chondrogenesis studies, differentiation is induced under serum-free media conditions in the presence of TGF-β3, a known inhibitor of matrix mineralization (Huang et al., 2010). In this micromass culture system, cellular condensation occurred at 1 day after TGF-β3 treatment and a spherical formation was confirmed at 2 days after (Fig. 2A). Consistent with this observation, the degree of precartilage condensation as assessed by PNA binding was significantly increased in TGF-β3 treated micromass (Fig. 2B). After 2 days, micromass cultures were stained with Alcian blue to visualize GAG content (Fig. 2C).
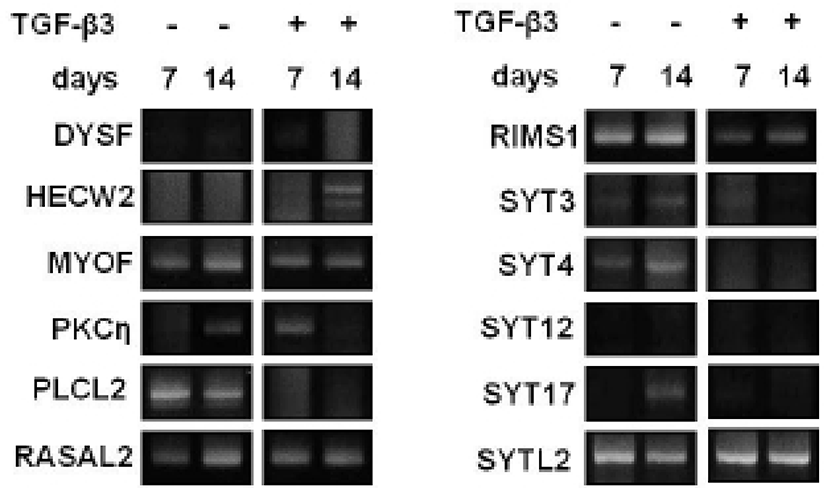
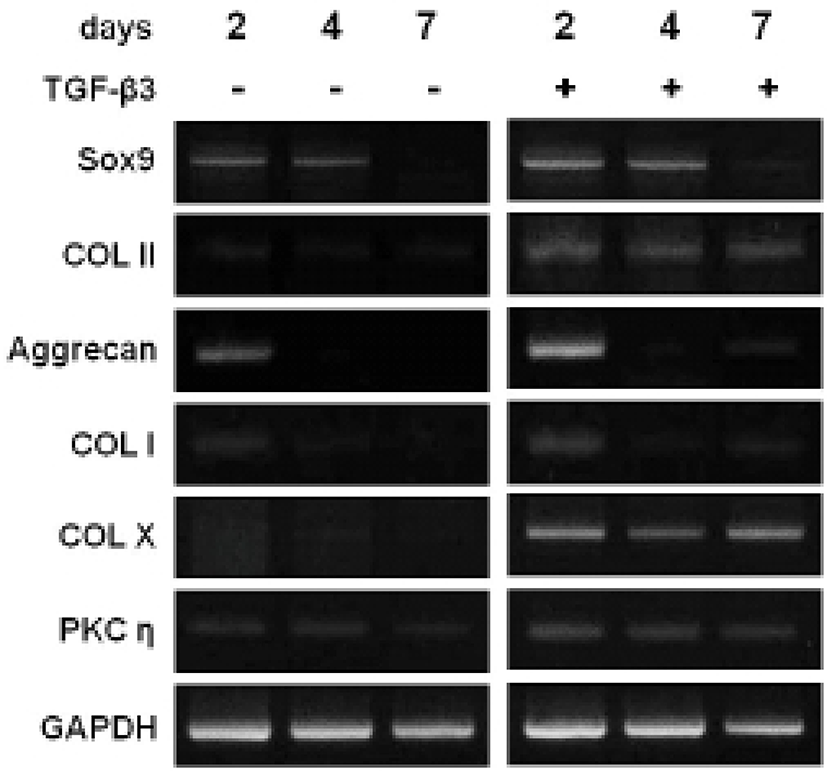
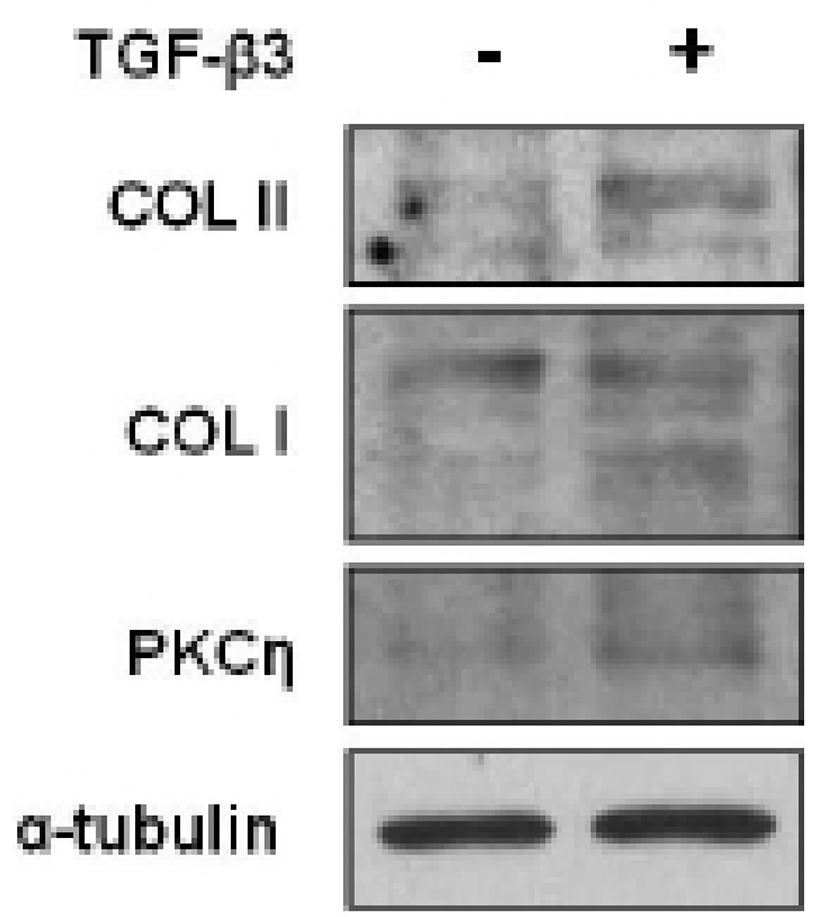
To evaluate which selected gene is closely related to the chondrogenesis of hMSC, gene expressions were analyzed by RT-PCR. Among 12 C2 domains, domain of PKCη showed similar expression pattern to COL II during TGF-β3 treatment (Fig. 3). The gene expression of Sox9, COL II, aggrecan, and PKCη strongly increased by day 2 of culture with TGF-β3, and expression remained until day 7 (Fig. 4). The levels of COL II and PKCη protein were also increased by TGF-β3 (Fig. 5). COL I, which signifies inadequate differentiation, did not show a significant increase in protein level (Fig. 5).
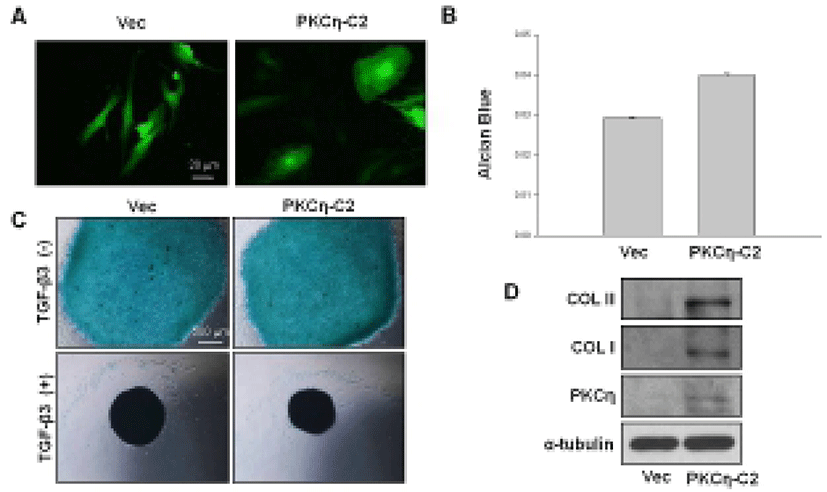
The C2 domain of PKCη is thought not to affect substrate-specificity of the kinase but instead aid in its localization to membranous systems (Littler et al., 2006). PKCη overexpression induces G1 arrest and differentiation in keratinocytes. In addition to epithelial cells, recent studies revealed that PKCη acts as a key regulator in early B-cell development (Kashiwagi et al., 2002). To test whether PKCη-C2 domain functions in chondrogenesis, hMSCs were infected with Adv-vec or Adv-PKCη-C2 domain and cultured in monolayer for 7 days. Although high-density cell culture environment is pivotal for the chondrogenic differentatiation of hMSCs, PKCη-C2 domain induced morphological change of hMSCs from a characteristic fibroblast-like morphology to a large round shape (Fig. 6A). In addition PKCη-C2 domain markedly induces chondrocyte matrix formation after 7 days of monolayer culture, as shown by Alcian blue staining of the cartilaginous matrix (Fig. 6B). Chondrogenic effect of PKCη-C2 domain was further confirmed in adenovirus infected MSCs micromass model by assessing spherical size and chondrocyte matrix formation (Fig. 6C). The level of a chondrogenic protein, COL II was also increased by PKCη-C2 domain (Fig. 6D). PKCη-C2 domain also increased chondrogenic differentiation in micromass model.
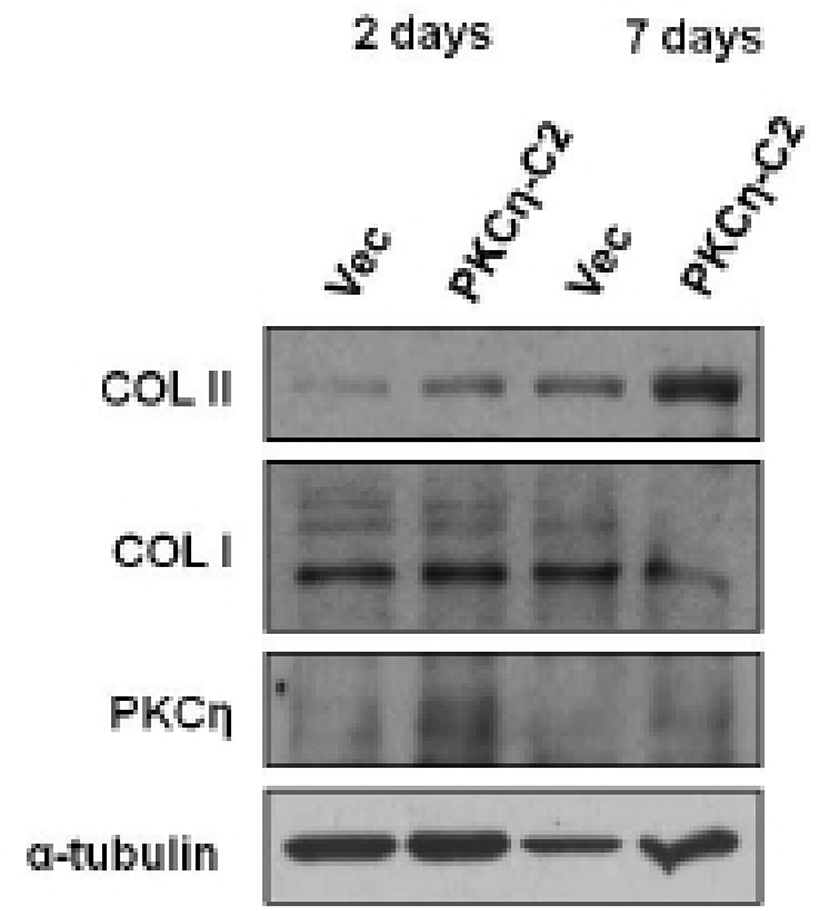
As shown in Fig. 6D, COL II gene expression was affected by PKCη-C2 domain. To investigate further the roles of PKCη-C2 domain in COL II gene expression, PKCη-C2 domain was overexpressed in NHFB cells. The COL II protein expression was increased by PKCη-C2 domain and this effect was more evident according to time (Fig. 7).
DISCUSSION
Natural chondrogenesis is a well-coordinated developmental differentiation program that leads to permanent articular cartilage in the joint or to transient cartilage during endochondral bone formation (Muir, 1995; Shum & Nuckolls, 2002; Csaki et al., 2008; Chen et al., 2009). Generation of stable hyaline cartilage from MSCs is currently still a challenge. Although chondrogenesis and cartilage formation are achieved, it eventually leads to terminal differentiation of chondrocytes instead of the production of stable hyaline cartilage (Pelttari et al., 2008; Huang et al., 2010). Further, the tissue-engineered cartilage construct is not stable when it is implanted in vivo but mineralizes (Pelttari et al., 2006). A better knowledge of mechanisms determining chondrocyte differentiation and terminal differentiation is therefore crucial to control the chondrogenic differentiation of MSCs. In the present study, it was demonstrated that PKCη-C2 domain regulates COL II gene expression and TGF-β3-induced chondrogenic differentiation.
Chondrogenic differentiation was achieved following the direct chondrogenic differentiation in vitro micromass protocol (Wang et al., 2005; Woods et al., 2007; Jin et al., 2010). The production of specific chondrogenic markers were measured at 2, 4, 7, and 14 days of culture in chondrogenic medium. The quantitative RT-PCR analysis measured the expression of COL I, COL II, and COL X in the spheroid formed during the chondrogenic process. Because spheroids first appear in this study after 2 days in culture in differentiation medium, the measurement of the expression of COL I, COL II, and COL X in the spheroids was began at 2 days. The expression of COL I, COL II, and COL X was found as early as 2 days of chondrogenic differentiation. These results are coincident with previous reports (Wang et al., 2005; Woods et al., 2007; Jin et al., 2010). COL II and COL X both increased their expression over time.
It is known that during the differentiation process of MSCs, one or several intracellular chemical cascades are modified influencing the ultimate commitment of the cell (de Crombrugghe et al., 2000). However, it remains unclear how each individual pathway affects the differentiation program of the cells and how manipulation of these pathways could lead to more efficient differentiation protocols.
PKC is a family of related protein kinase, which includes at least 10 different isoforms in mammalian cells (Newton, 1997). They play important roles in the transduction of signals coupled to receptor-mediated hydrolysis of membrane phospholipids. The mammalian isoenzymes can be classified into three groups according to their structure and cofactor regulation. The first group includes the classical isoforms (α, βI, βII, and γ), which function is regulated by calcium, acidic phospholipids, and diacylglycerol. The second group corresponds to the novel PKCs (δ, ε, η, and θ), which are activated by acidic phospholipids and diacylglycerol in a calcium-independent manner. These two groups contain in their regulatory regions both conserved C1 domains responsible for sensing diacylglycerol, and C2 domains responsible for sensing Ca2+ and/or acidic phospholipids at different subcellular compartments. The third group comprises the atypical PKC isoforms (ξ, τ /λ), which are not regulated by diacylglycerol or by calcium (Nishizuka, 1992; Rosse et al., 2010).
The C2 domains of classical and novel PKC play a important role in decoding signals, which trigger the translocation of these enzymes to the plasma membrane and/or other membrane (Littler et al., 2006; Corbalán-García & Gómez-Fernández, 2010). Although the C2 domains of novel PKCs were supposed to play only a secondary role with respect to the C1 domain in the activation process of these enzymes, new insights reveal that these C2 domains may also receive regulatory inputs and play an important role in the localization and activation of these enzymes (Nishizuka, 1992; Newton, 1997; Corbalán-García & Gómez-Fernández, 2010; Rosse et al., 2010).
The novel PKCη isoform has a unique tissue distribution and is primarily expressed in epithelial tissue and cells undergoing high turnover (Kashiwagi et al., 2002). It is implicated in diverse cellular functions, including a role in terminal differentiation, proliferation, and secretion (Shtutman et al., 2003; Lampasso et al., 2006; Adhikary et al., 2010; Lee et al., 2010). Recent studies suggest that PKCη has a special role in response to stress and regulation of apoptosis (Rotem-Dai et al., 2009). However the function of the PKCη in chondrogenesis is not well understood.
In this study, PKCη gene expression increased during TGF-β3-induced chondrogenic differentiation. In addition, PKCη-C2 domain induced the gene expression of COL II in monolyer and micromass culture. The molecular mechanisms activated in MSCs, leading to the increased expression of PKCη, are currently not understood. Chondrogenesis might be a product of orchestration of related genes as shown in the TGF-β3/bone morphogenic protein 2-induced chondrogenesis of MSC (Sang et al., 2014). However, introduction of one protein into human amniotic fluid-derived stem cells changed the pluripotency (Jo et al., 2010). Furthermore, PKCδ gene silencing with shRNA caused a severe reduction in cartilage formation (Matta et al., 2011). Although it seems not to enough, only PKCη-C2 domain can induce chondrogenesis via influencing the Ca2+-related signaling.
In summary, the present study demonstrates interaction between PKCη-C2 domain and the COL II gene expression, providing new insights into the possible links of PKCη to the chondrogenesis. Further studies are needed for the elucidation of the molecular mechanisms involved.