
INTRODUCTION
At the cellular level, obesity is caused by an increase in the number and size of adipocytes in adipose tissue (Spiegelman and Flier, 1996). Adipocytes accumulate excessive concentrations of triglycerides; this interferes with the signaling pathways involved in the normal metabolism of adipocytes, causes insulin resistance, and may even lead to metabolic syndrome abnormalities (Kahn and Flier, 2000). Obesity may be further characterized as a state of chronic low-grade inflammation marked by the infiltration of macrophages into adipose tissue (Weisberg et al., 2003; Xu et al., 2003). Interactions between hypertrophic adipocytes and infiltrated macrophages aggravate inflammation and lead to insulin resistance in obese adipose tissue (Dandona et al., 2004; Fernández-Real and Ricart, 2003).
Adipose tissue macrophages are a major source of pro-inflammatory cytokines, which play important roles in chronic inflammatory responses in obese adipose tissue (Itoh et al., 2011). For example, tumor necrosis factor (TNF)-α directly decreases insulin sensitivity and increases lipolysis in adipocytes (Hotamisligil et al., 1995; Zhang et al., 2002). Interleukin (IL)-6 leads to hypertriglyceridemia in vivo by stimulating lipolysis and hepatic triglyceride secretion (Nonogaki et al., 1995). In addition, induced nitric oxide synthase (iNOS) and TNF-α are required for the development of obesity-induced insulin resistance in mice (Perreault and Marette, 2001). The pro-inflammatory cytokines produced by both adipocytes and recruited macrophages might synergistically decrease insulin sensitivity (Dandona et al., 2004; Hotamisligil et al., 1995).
Tangeretin, a type of polymethoxyflavone (PMF), is found in citrus plants and exerts anti-inflammatory, anti-cancer, and anti-obesity effects (Chen et al., 2007; Choi et al., 2007; Kim et al., 2012; Morley et al., 2007). It was used in traditional medicine for treatment of various diseases. Also, tangeretin modulates glucose metabolism in streptozotocin induced diabetic rats (Sundaram et al., 2014). However, little is known about the effects of tangeretin on the inflammatory responses and insulin resistance caused by direct interactions between adipocytes and macrophages. In the present study, we investigated the effects of tangeretin on inflammatory changes and glucose uptake in a coculture of hypertrophic adipocytes and macrophages.
MATERIALS AND METHODS
We obtained 3T3-L1 preadipocyte cells and RAW 264.7 murine macrophage cells from the American Type Culture Collection (Rockville, MD, USA) and the Korea Cell Line Bank (Seoul, South Korea), respectively. The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Gibco BRL, Gaithersburg, MD, USA), which contained 1 % penicillin/streptomycin (PS) (100 units penicillin/mL and 100 pg streptomycin/mL) and was supplemented with either 10% bovine calf serum (BCS, Gibco, NY, USA) or 10% fetal bovine serum (FBS, Gibco, NY, USA), at 37℃ in a 5% CO2 atmosphere. To prepare hypertrophic adipocytes, post-confluent preadipocytes (designated as day 0) were cultured until confluent in differentiation medium [DMEM containing 1% PS, 10% FBS, 0.5 mM 3-isobutyl-1-methylxanthine (IBMX), 1 µM dexamethasone, and 1 µg/mL insulin] for 2 days. The cells were then cultured for 2 more days in DMEM containing 1% PS, 10% FBS, and 1 µg/mL insulin. Thereafter, the hypertrophic 3T3-L1 cells with larger lipid droplets were prepared in post-differentiation medium (DMEM containing 1% PS and 10% FBS) for 14 days, which was replaced every 2 days. Co-culturing of adipocytes and macrophages was performed in accordance with a previously described protocol (Jeon et al., 2015; Shin et al., 2016). RAW 264.7 macrophages were plated in culture dishes containing serum-starved DMEM. Then, they were plated onto a hypertrophic 3T3-L1 monolayer and the cocultured cells were incubated in serum-starved DMEM for 24 h. Equal numbers of RAW 264.7 macrophages and 3T3-L1 adipocytes were cultured separately as controls. After 24 h, tangeretin (Wako Pure Chemical Industries, Osaka, Japan) was added to the cocultured cells at various concentrations for an additional 24 h. The supernatants and cells were collected and stored at −20℃ until use. Cell cytotoxicity was determined by lactate dehydrogenase (LDH) release assay (Takara Shuzo Co., Otsu, Japan).
The amount of nitrite was determined by colorimetric assay. Briefly, 100 µL of cell culture medium was mixed with an equal volume of Griess reagent (1% sulfanilamide and 0.1% naphthylethylenediamine in 5% phosphoric acid) and incubated at room temperature for 10 min. The absorbance at 540 nm was recorded using a microplate reader (BioTek instrument Inc., Winooski, VT, USA). The nitrite concentration was determined by extrapolation from a sodium nitrite standard curve.
Cells were homogenized with lysis buffer [1×RIPA (Upstate Cell Signaling Solutions, Lake Placid, NY, USA), 1 mM phenylmethylsulfonyl fluoride, 1 mM Na3VO4, 1 mM NaF, 1 µg/mL aprotinin, 1 µg/mL pepstatin, and 1 µg/mL leupeptin] and incubated on ice for 1 h. Cell lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene difluoride membranes. These membranes were then blocked with a solution of 0.05% Tween 20/Tris-buffered saline containing 5% non-fat dry milk for 1 h at room temperature. Antibody against iNOS was obtained from Calbiochem (San Diego, CA, USA), and antibody against COX-2 was obtained from BD (Mountain View, CA, USA). Anti-phospho-AKT and anti-insulin receptor substrate (IRS) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Antibodies against phospho-IRS and AKT were obtained from Cell Signaling Technologies (Beverly, MA, USA). After incubation overnight at 4℃ with the indicated primary antibody, the membranes were incubated with the horseradish peroxidase-conjugated secondary antibody (Jackson ImmunoResearch, West Grove, PA, USA) for 1 h at room temperature. Immunodetection was carried out using the enhanced chemiluminescence (ECL) Western blotting detection reagent (Amersham Biosciences, Piscataway, NJ, USA).
Total RNA was isolated from cells using Trizol reagent (Molecular Research Center Inc., Cincinnati, OH, USA). RNA concentrations were measured from the absorbance at 260 and 280 nm. Complementary DNA (cDNA) synthesis was performed using a Maxime RT PreMix Kit (Intron Biotechnology, Seoul, South Korea), in accordance with the manufacturer’s protocol. The following primers were used for real-time RT-PCR analyses: IL-6, 5′-GTA CTC CAG AAG ACC AGA GG-3′ and 5′-TGC TGG TGA CAA CCA CGG CC-3′; IL-1β, 5′-CAG GAT GAC ATG AGC ACC-3′ and 5′-CTC TGC AGA CTC AAA CTC CAC-3′; TNF-α, 5′-TTG ACC TCA GCG CTG AGT TG-3′ and 5′-CCT GTA GCC CAC GTC GTA GC-3′; and β-actin, 5′-AGG CTG TGC TGT CCC TGT AT-3′ and 5′-ACC CAA GAA GGA AGG CTG GA-3′. The levels of IL-1β, IL-6, TNF-α, and β-actin mRNAs were measured using a Chromo 4TM Real-Time PCR Detector (Bio-Rad Laboratories Inc., Hercules, CA, USA). For relative quantification, the reactions were performed using 2× iQTM SYBR Green Supermix (Bio-Rad). The PCR amplification was performed for 44 cycles, each lasting 20 s at 95℃ and 65℃, and 30 s at 72℃, with initial denaturation at 95℃ for 3 min. Cytokine mRNA levels were compared after correction using the β-actin mRNA level as an internal standard. Expression level was analyzed by gene expression analysis using the Chromo 4 instrument (Bio-Rad Laboratories Inc., Hercules, CA, USA).
Glucose uptake was detected using fluorescent 2-[N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl) amino]-2-deoxyglucose (2- NBDG; Invitrogen, Carlsbad, CA, USA). Briefly, hypertrophic 3T3-L1 adipocytes grown in 12-well plates were washed twice with serum-free DMEM and incubated in DMEM containing 0.2% bovine serum albumin for 4 h. Then, RAW 264.7 cells were added to 3T3-L1 adipocytes and incubated for a further 24 h before treatment with tangeretin. After 24 h, the cells were washed with Krebs-Ringer-Hepes (KRH) buffer (Boston BioProducts, Inc., Ashland, MA, USA) and treated with tangeretin (100 µM), insulin (100 nM), and 2-NBDG (100 µM) for 15 min at room temperature in dark condition. Glucose uptake was terminated by washing the cells three times with cold PBS. The cells were then lysed and harvested using 1% Triton X-100. The fluorescence intensity was measured at an excitation of 485 nm and emission of 520 nm using a fluorescence micro-reader (Fluostat Optima; BMG Labtech, Germany).
RESULTS AND DISCUSSION
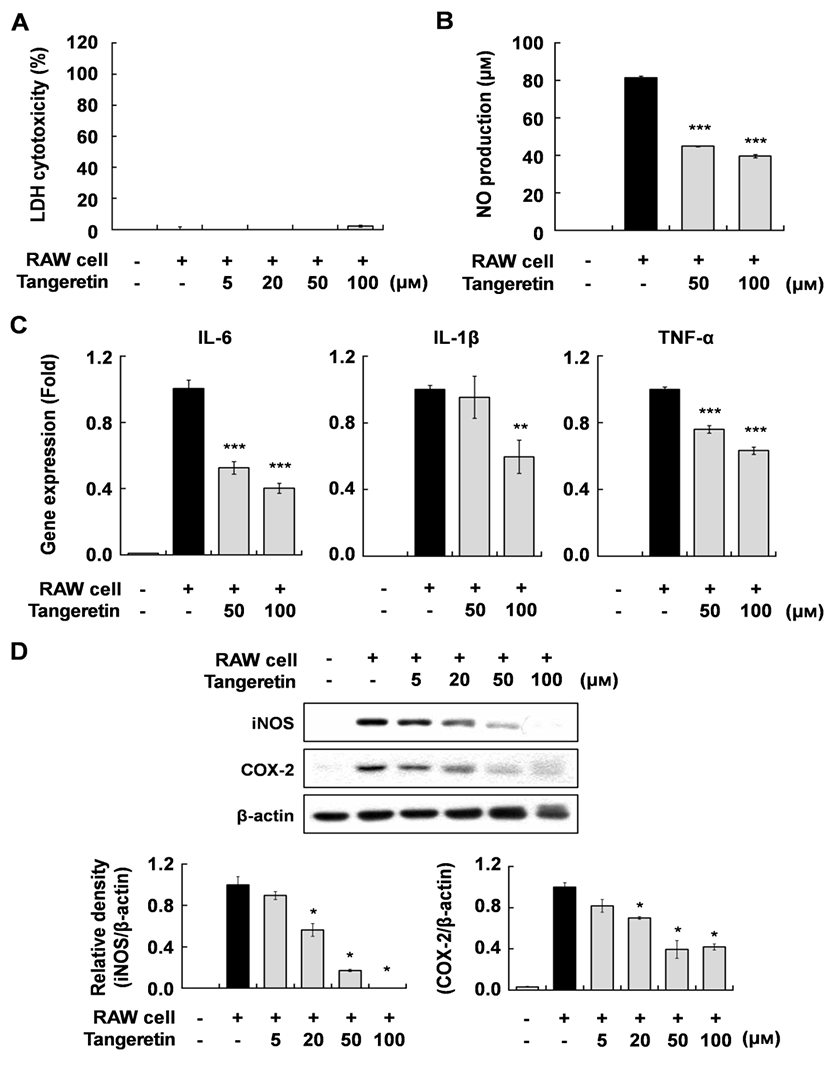
To investigate the effect of tangeretin on inflammatory changes in cocultured hypertrophic adipocytes and macrophages, we first evaluated the cytotoxicity of tangeretin in this system using MTT and LDH assays. Tangeretin did not affect the cytotoxicity up to a concentration of 100 µM (Fig. 1A). When the hypertrophic 3T3-L1 cells were cocultured with RAW 264.7 cells for 24 h and incubated for another 24 h, inflammatory mediators were markedly increased. However, tangeretin treatment significantly decreased NO production and the expression of IL-6, IL-1β, TNF-α mRNA, and iNOS and COX-2 proteins in a dose-dependent manner (Fig. 1B, C, D). These results indicate that tangeretin attenuates the inflammatory response caused by interaction between hypertrophied adipocytes and macrophages. Pro-inflammatory cytokines, including IL-6, IL-1β, and TNF-α, have been reported to have direct effects on adipose tissue cellular metabolism and insulin resistance (Luca and Olefsky 2008; Suganami et al., 2005; Wang et al. 2008). These anti-inflammatory effects of tangeretin in cocultured hypertrophic adipocytes and macrophages might improve insulin resistance in obese adipose tissue.
Therefore, we investigated the effects of tangeretin on glucose uptake and the insulin signaling pathway in cocultured hypertrophic adipocytes and macrophages. When the hypertrophic adipocytes were cocultured with macrophages, glucose uptake was decreased compared with that in culture without macrophages, indicating that insulin resistance was induced in the coculture of hypertrophic adipocytes and macrophages (Fig. 2A). Tangeretin treatment significantly improved glucose uptake, and co-treatment with tangeretin and insulin further enhanced glucose uptake, in the coculture of hypertrophic adipocytes and macrophages (Fig. 2A). In typical insulin signaling, the binding of insulin to its receptor induces insulin receptor substrate (IRS) phosphorylation, initiating insulin signal transduction. The phosphorylation of IRS activates the phosphatidylinositol-3-kinase (PI3K)/Akt pathway, which in turn stimulates the uptake of glucose into cells by inducing translocation of glucose transporters to the plasma membrane (Kanzaki, 2006). Tangeretin did not affect the phosphorylation of either IRS or Akt (Fig. 2B), suggesting that the effect of tangeretin on glucose uptake might be related to insulin sensitivity.
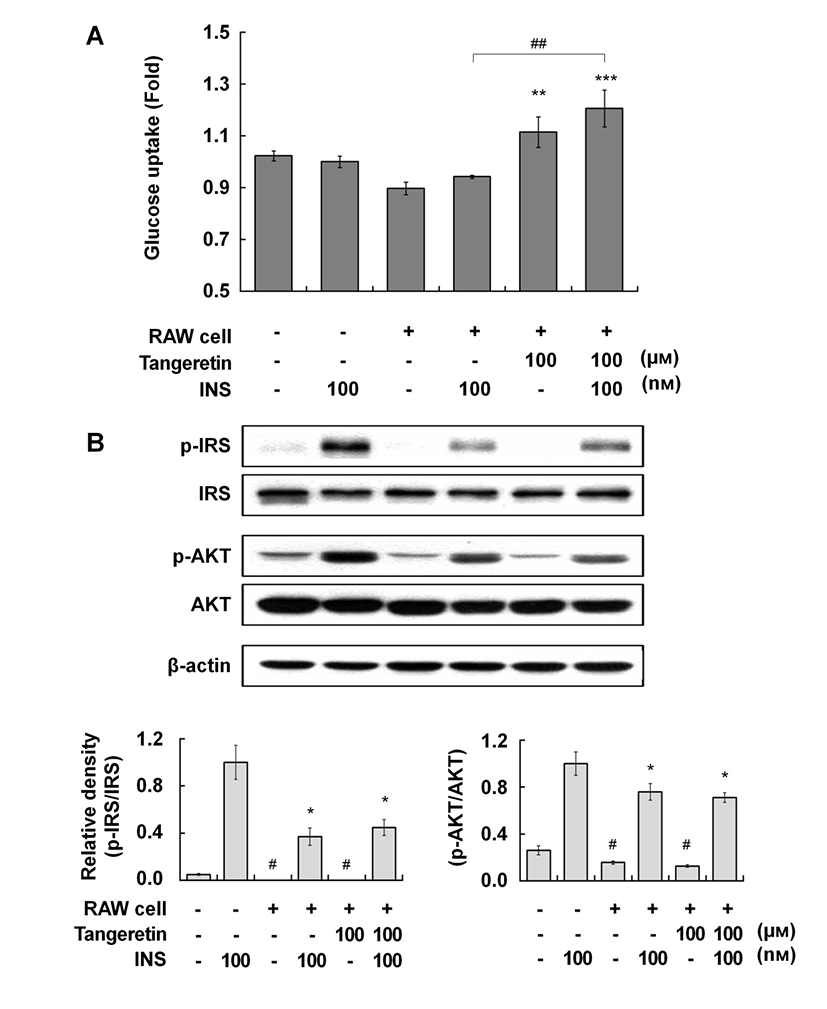
Several signaling cascades have been shown to be involved in glucose uptake in adipocytes. It has been reported that tangeretin increased glucose uptake via the PI3-K/Akt and PKA/CREB pathways in murine adipocytes (Onda et al., 2013). Kang et al. (2012) reported that dietary supplementation of immature Citrus sunki peel extracts, which contain high levels of PMFs, including tangeretin, increased the phosphorylation of AMP-activated protein kinase (AMPK) in epididymal adipose tissue in high-fat-diet-induced obese mice. Several phytochemicals improve insulin-independent glucose uptake by stimulating the activity of skeletal muscle AMPK (Kim et al., 2012; Zygmunt et al., 2010). The activation of AMPK inhibits the synthesis of pro-inflammatory cytokines, such as IL-6, IL-1β, and TNF-α, in macrophages (Bijland et al., 2013). Thus, the effect of tangeretin on glucose uptake in cocultured hypertrophic adipocytes and macrophages might be partly exerted via an insulin-independent AMPK pathway, although further study is required to uncover the precise molecular action of tangeretin. In summary, this study suggests that tangeretin exerts beneficial effects by attenuating inflammatory changes and increasing glucose uptake in obese adipose tissue.