
INTRODUCTION
Various nucleation-promoting factors (NPFs), including N-WASP (Yi et al., 2011), WAVE2 (Sun et al., 2011), and junction-mediating and regulatory protein (JMY) (Zuchero & Mullins, 2009; Lin, 2014, 2015), in addition to the actin nucleator, play essential roles in basic cellular processes, including migration, adhesion, division, membrane trafficking (Lin et al., 2014), oocyte maturation (Lin et al., 2015), and embryo development (Lin et al., 2015). JMY was originally identified as a p53 coactivator, which localizes to the nucleus during DNA damage (Coutts & La Thangue, 2007), and binds to and activates the Arp2/3 complex in response to Rho family GTPase signaling that are essential components for dynamic actin reorganization, plays a pivotal role in regulating the biochemical and cytoskeletal pathways relevant to cell migration. Previous studies have shown that JMY forms a complex with strap (stress-responsive activator of p300) and p300, which recruits PRMT5 (protein arginine methyltransferase 5) into a coactivator complex that drives the p53 response (Coutts et al., 2007; Cho & Lee, 2013; Lin et al., 2015; Wang et al., 2013).
Recently, it was showed that the clustered regularly interspaced short palindromic repeats (CRISPR) system could be harnessed in vivo (Jao & Chen, 2013; Guo et al., 2014). The CRISPR/Cas9 system could be efficiently used to generate knock out or knock in organisms via zygotic injections of Cas9 and single guide RNA (sgRNA) in many organisms, including the mouse (Cho et al., 2013; Wang et al., 2013), rat (Coutts et al., 2007), zebrafish (Guo et al., 2014; Hai & Zhou, 2014), nematode (Coutts et al., 2007), frog (Wang et al., 2013), pig (Cho et al., 2013), and monkey (Jao et al., 2013), indicating the versatility and universality of the CRISPR/Cas9 system in genome editing.
Considering the dual roles of JMY as a DNA damage responsive element and as an actin nucleation promoting factor by regulating the NPF-Arp2-actin pathway in the oocyte and embryo of pig (Lin et al., 2014; Lin et al., 2015), we postulated that JMY could be involved in the early embryonic development in mouse.
In this study, we investigated the roles of JMY by the CRISPR/Cas9 system gene targeting in mice, and showed that it is essential for the early embryonic development in mice. We showed that JMY is a maternal protein that is present in embryos. Though the embryos lacking this protein did not display a reduced ability for the first cleavage, a complete arrest at the blastocyst stage was observed in the embryo.
MATERIALS AND METHODS
A previously described plasmid (pCAG-T3-hCAS-pA) (Cho et al., 2013), which contained human codon optimized Cas9 plasmid with T3 promoter, was obtained from Addgene (41815) and used to generate Cas9 mRNA. Briefly, the plasmid was linearized by digestion with SphI and transcribed using an mMessage Machine T3 Kit (Life Technologies, Foster City, CA, USA). After transcription, the template DNA was removed by treatment with Turbo-DNase (Life Technologies, Foster City, CA, USA). The resulting transcripts were purified by phenol-chloroform extraction and isopropanol precipitation, and stored at −80°C until further use. Guide sequences of sgRNAs corresponding to JMY were designed using the CRISPR Design Tool (Cho et al., 2013) (http://crisp.mit.edu). Forward PCR primers containing the T7 promoter, guide sequences, and portions of the Cas9 handle were hybridized with a reverse PCR primer containing the Cas9 handle and Streptococcus pyogenes terminator sequences, and then amplified by PCR using the Phusion DNA polymerase (Thermo Fisher Scientific, Waltham, MA, USA). The T7 promoter primer and the terminator primers are listed in Table S1. The resulting PCR products (123 bp) were purified by gel extraction and used as templates for in vitro transcription by using the T7 Mega-shortscript kits (Life Technologies, Foster City, CA, USA), followed by purification with phenol-chloroform extraction, as performed with the Cas9 mRNAs.
Mice belonging to the ICR strain were used for the experiments. Animal care and handling was conducted in accordance with the policies regarding the care and use of animals issued by the ethical committee of the Department of Animal Science, Chungbuk National University, Korea. Ovaries from 6- to 8-week-old female mice were removed and cut into pieces using a razor blade. The tissue was washed in M2 medium (Sigma; St. Louis, MO, USA). Female Mice were injected with 10 IU pregnant mares serum gonadotropin (PMSG) by nintraperitoneal injection, and then with 10 IU human chorionic gonadotropin (hCG) after 2 days of PMSG priming. To collect embryos in vivo, estrous females were mated with the males; one-cell (pronuclear stage) and two-cell stage embryos were collected from the hormone-injected mice after 27–28 h. Microinjections were performed within 1 h of collection, using a microinjector (Eppendorf, Hamburg, Germany). After injections, the embryos were transferred in groups of 15–30 in KSOM/AA medium (60 μL) under mineral oil (Dow Corning, UK) at pre-equilibrated (5% CO2, 37°C) growth conditions, and placed in a water-jacketed incubator (Kendro, UK) at 37°C.
Total RNA was isolated from frozen embryos with the Dynabeads mRNA direct kit (Dynal Asa, Oslo, Norway) and reverse transcribed into cDNAs with oligo (dT)12–18 and SuperScript II reverse transcriptase (Invitrogen, Grand Island, NY, USA). RT-qPCR was performed with the DyNAmo HS SYBR Green qPCR kit (FINNZYMES, Helsinki, Finland), and post-PCR analysis was done in a CFX96 real-time qPCR system (Bio-Rad, Hercules, CA, USA). The thermal cycling conditions for the RT-qPCR were as follows: 94°C for 30 s, followed by 40 cycles at 94°C for 30 s, 60°C for 30 s, and 72°C for 25 s. A final extension at 72°C for 5 min was included at the end of the run. For normalization, peptidylprolyl isomerase A (Ppia) RNA was used as an internal control. Briefly, for each independent experiment, mRNA was extracted from 20 embryos from each stage.
Antibodies to detect Oct4 (sc-8628), JMY (sc-13020, Santa Cruz Biotechnology, Inc. CA, USA), Cdx2 (MU392A-UC, BioGenex Laboratories Inc. San Ramon, CA, USA)), and GAPDH (ab9484, Abcam Inc. Cambridge, MA, USA) were used for the immunoassays. For immunocytochemistry, the oocytes were fixed in 4% paraformaldehyde for 30 min, and then treated with 0.5% Triton X-100 for 20 min. After blocking in 1% bovine serum albumin (BSA) for 1 h, the oocytes were incubated with the primary antibody (1:50 dilution) at 38°C overnight. After five washes in phosphate buffered saline (PBS) containing 0.05% Tween 20 (PBST), oocytes were incubated with tetramethylrhodamine (TRI-TC)-conjugated secondary antibody (Life Technologies, Carlsbad, CA) diluted 1:100 times in PBST for 1 h. After four washes in PBST, oocytes were incubated for 15 min with Hoechst 33342 dye (0.5 μg/mL), prepared in PBS. Oocytes were mounted on glass slides and examined by laser scanning confocal microscopy (models LSM 510 and 710 META, Zeiss, Oberkochen, Germany).
For western blotting, mouse embryos were collected in SDS sample buffer and incubated for 5 min at 100°C. Proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes, which were blocked in Tris buffered saline supplemented with Tween 20 (TBST) containing 5% BSA for 2 h, and then incubated overnight at 4°C with the primary antibody (1:500 dilution). After three 10-min washes in TBST, the membranes were incubated for 1 h at 37°C with a peroxidase-conjugated secondary antibody (1:2,000 dilution). Membranes were processed using the SuperSignal West Femto maximum sensitivity substrate (ThermoFisher Scientific, Waltham, MA).
All the percentage data were subjected to arcsine transformation before statistical analysis. The general linear models (GLM) procedure in the SAS program (SAS Institute Inc., Cary, NC, USA) was used to analyze the data. Differences with p values less than 0.05 were considered statistically significant.
RESULTS
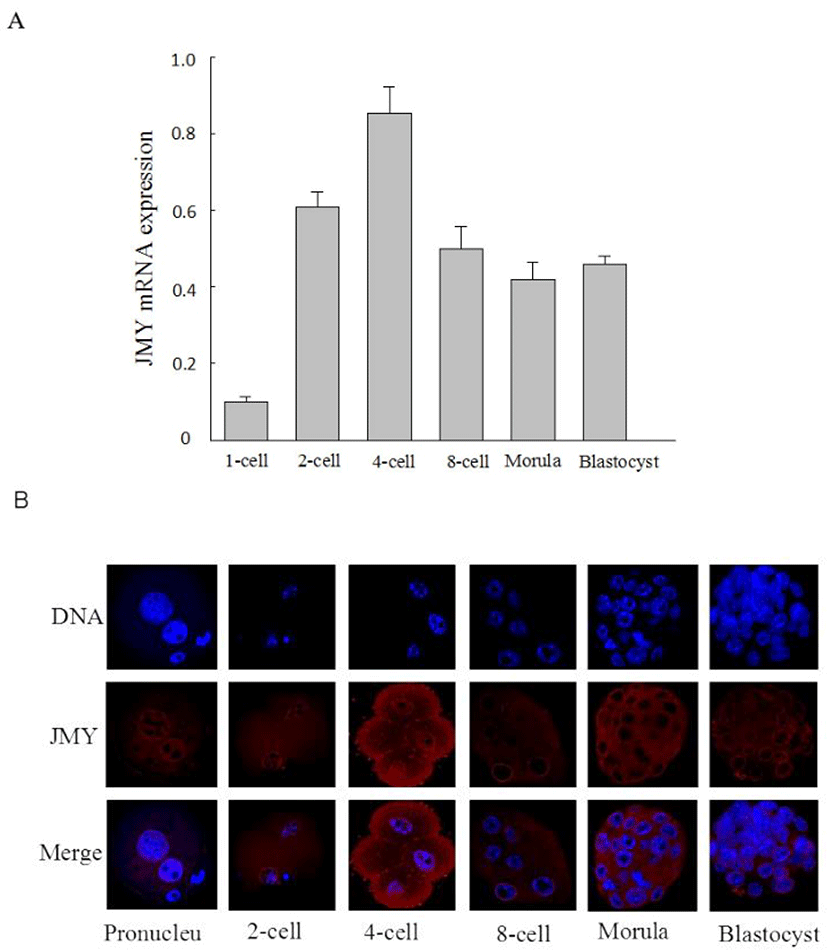
We investigated the expression of JMY mRNA and the subcellular localization of JMY protein in the mouse embryos. JMY mRNA expression was increased significantly after the 2-cell stage, and was maintained at the same level at the 4-cell stage, and then, decreased from the 8-cell to the blastocyst stage (Fig. 1A). We also determined JMY localization by immunofluorescent staining (Fig. 1B). JMY was predominantly located in the nucleus of the pronuclear stage embryos; at the 2-cell stage, it distributed in the plasma surrounding the nucleus; at the 4-cell stage, it was seen in the nucleus and cytoplasm; at morula and blastocyst stages, JMY protein selectively enriched in the nuclear membrane.
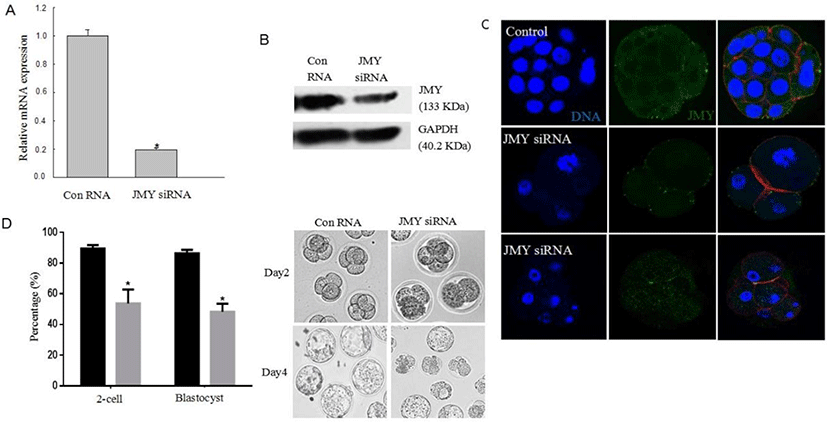
We injected JMY-specific small interfering RNA (siRNA) to assess the function of JMY during mouse embryo development. As shown in Fig. 2A–C, we confirmed the knockdown of JMY in the embryos. We observed that most control embryos developed to the blastocyst stage. However, in the JMY-knockdown embryos, majority of them were arrested at the 2-cell stage. In addition, embryo development up to the blastocyst stage was significantly higher (83.4±2.3%) in the controls than that in the JMY-knockdown embryos (44.7±2.2%; p<0.05) (Fig. 2D). Furthermore, zygotes with knockdown of JMY gene frequently exhibited an abnormal cleavage, as compared to the controls (Fig. 2C).
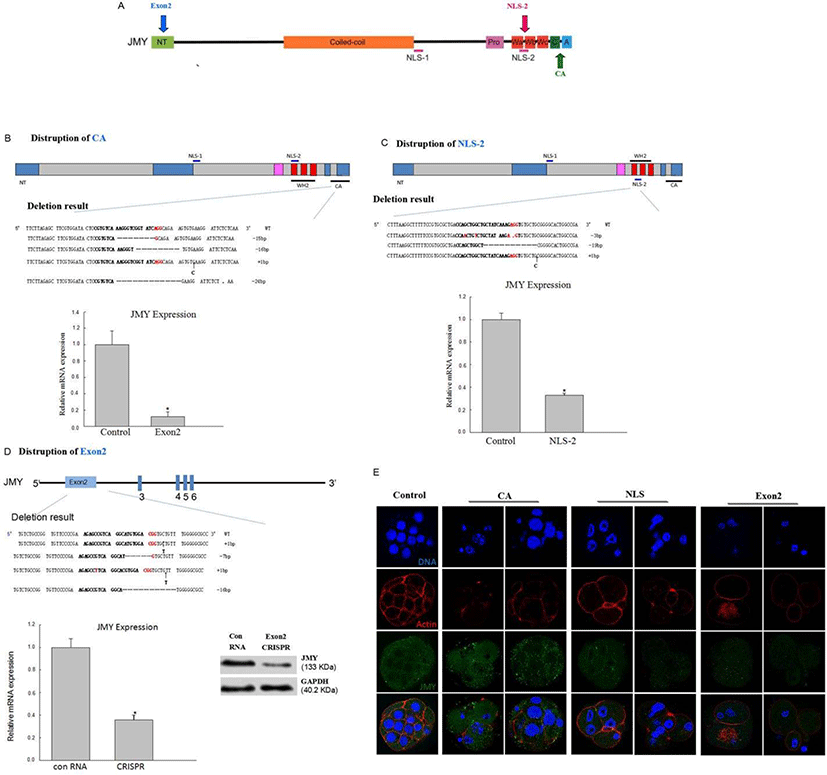
To explore the function of the JMY gene, we chose to knockdown exon 2, CA, and nuclear localization signal domain (NLS-2) regions of the JMY gene (Fig. 3A). We injected 400 and 200 ng/μL Cas9 mRNA and 20 and 40 ng/μL sgRNA for knocking down exon 2, CA, and NLS-2 regions of the JMY gene into the 1-cell stage zygotes of mouse; while we only injected the Cas9 mRNA in the controls. We checked the mutation status by pooling genomic DNA collected from the embryos by using the guide sequences (Fig. 3B, C, D). We found that the location of JMY in CA, exon 2, and NLS-2 group was different from that in the control group (Fig. 3E). As shown in Fig. 4A and 4B, embryos lacking the JMY gene had severely reduced blastocyst formation. We generated JMY-knockout mice embryos by deleting exon 2 of the JMY gene, which displayed reduced ability for the first cleavage (Fig. 4B).
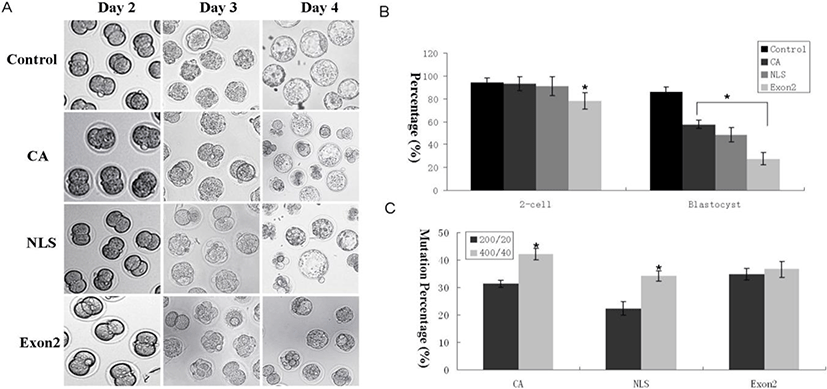
Initially, to determine the optimal concentration of Cas9 mRNA for gene targeting in vivo, we microinjected various amounts of Cas9-encoding mRNA (200 and 400 ng/μL) with JMY-targeting sgRNA at constant concentrations (20 and 40 ng/μL) into the 1-cell pronuclear stage of the mouse embryos. As expected, higher concentrations of Cas9 mRNA led to more efficient gene disruption (Fig. 4C). Nevertheless, even embryos injected with Cas9 mRNA (200 ng/μL), showed normal blastocyst development (Fig. 4A and 4B).
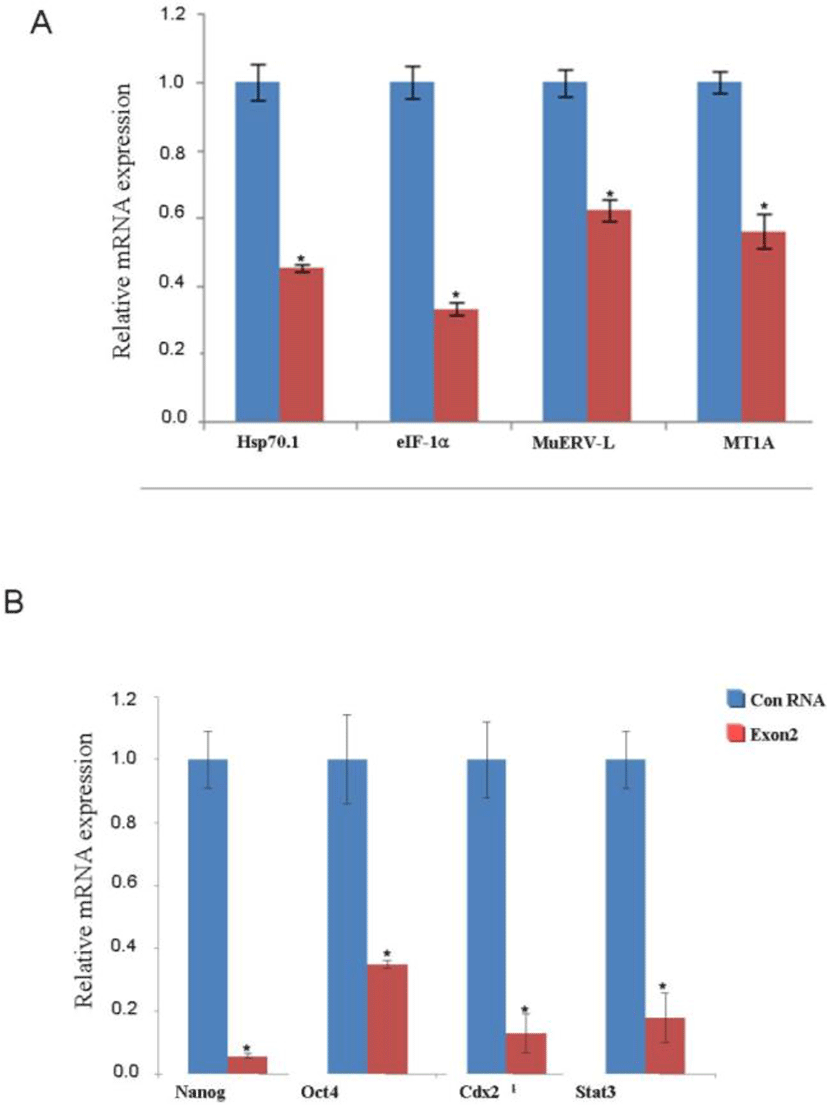
We hypothesized that JMY affects the expression of other development-related transcripts, including those of Nanog, Oct4, Sox2, and Stat3. We analyzed the levels of these mRNAs at 48 h after fertilization in the controls and JMY-exon 2 knockout group by using real-time qPCR. We found that the location of Cdx2 and Oct4 did not change in the JMY-exon 2 knockout group (Fig. 5A); however, there was a significant decrease in the mRNA levels of transcription- and zygotic genome activation (ZGA)-related genes in the JMY-exon 2 knockout group (Fig. 5B, C).
DISCUSSION
In this study, we investigated whether JMY was involved in the embryonic development of mouse, and found that JMY played important roles in regulating embryo cleavage and blastocyst formation. In addition, the generation of mice with specific mutations by CRISPR/Cas9-mediated gene targeting was efficient. We showed that knockdown of JMY gene by siRNA arrested the early embryonic development in mice.
To investigate the effectiveness of the CRISPR/Cas9 system in the functional characterization of genes involved in mammalian preimplantation development, we studied the function of JMY by gene targeting in mice zygotes. It was reported that actin monomers block JMY nuclear import at NLS-2 site (Zeng & Schultz, 2005). Therefore, we knocked out NLS-2 in this experiment to explore the potential function in embryo. In addition, we introduced indels in the exon 2, CA (Arp2/3 binding site), and NLS-2 site of the JMY gene via non-homologous end joining (NHEJ) repair mechanism to explore the JMY gene function. Our results showed that the introduction of a single sgRNA/Cas9 pair yielded high efficiency (30%–50%, depending on the guide RNA sequence) during genetic modification.
JMY was discovered as a unique actin assembly protein that nucleates actin both as a tandem-monomer-binding nucleator and as an NPF (Zeng & Schultz, 2005), and functions as a p300 coactivator in the p53 response (Shikama et al., 1999; Zuchero et al., 2009). It was reported that complete JMY gene was fully active as an actin nucleator and NPF in vitro (Coutts & La Thangue, 2009). In this study, we showed that deletion on NLS-2 site in the JMY gene altered JMY localization. As JMY has been reported to localize to both the nucleus and the cytosol or leading edge (Zuchero et al., 2009; Lin et al., 2015), previous studies have demonstrated that JMY accumulates in the nucleus upon DNA damage and relocates from the nucleus to the leading edge in migrating cells (Coutts et al., 2007; Lin et al., 2014). We also observed distribution of JMY between the nucleus and the cytosol in the early embryonic development in mouse. At normal conditions, JMY protein localized to the cytoplasm and nucleus; however, when NLS-2 site was deleted, redistribution of JMY occurred in the nucleus only. Clearly, more work is needed to determine the mechanisms that regulate JMY distribution between the nuclear and cytosolic compartments.
In addition to examining its role in zygote development, we also tested the function of JMY in embryos because it is highly expressed in the 2-4-cell stage during ZGA, indicating that it plays important roles in cell division, including embryo development (Sun et al., 2011).
Although the molecular machinery that regulates actin polymerization in neurons is not fully characterized, different actin nucleators, including the Arp2/3 complex (Zuo et al., 2017) and its NPFs N-WASP (Pinyol, 2007; Ceglia et al., 2017), WAVE1 (Firat-Karalar & Welch, 2011) and WAVE 2 (Hoeng et al., 2008), as well as the tandem actin-monomer-binding nucleator Cordon-bleu (Coutts et al., 2009) were shown to positively regulate the stages of neuronal development through their role in growth cone formation and dynamics. In contrast, we found that knockout of CA (Arp2/3 binding site) on JMY gene obviously affected blastocystformation. Based on our observation that JMY plays a negative role in the formation of the pronucleus, and JMY being a transcription-related protein, we further investigated its function in the 2-4-cell stage, when zygotic gene transcription became active. We found that JMY plays a role in embryo development by downregulating the expression of Nanog, Oct4, Cdx2, and Stat3, and not by affecting the pronucleus. Oct4 and Cdx2 localization was normal upon knockout of exon 2. One possibility was that JMY acts as an upstream effector of ZGA genes such as Hsp70.1, eIF-1A, mUerv-l and MTIA. Actin-nucleating proteins are regulated by small GTPases, protein–protein interactions, and/or posttranslational modifications, and it is likely that JMY is also regulated by similar mechanisms (Ceglia et al., 2017). Another intriguing question is why JMY combines two uniquely different cellular functions-transcriptional regulations and actin assembly in one protein. Given the emeging role of nuclear actin in transcription regulation (Ceglia et al., 2017), it can be speculated that JMY acts as a molecular link between the actin cytoskeleton and the transcriptional machinery. It will be important to examine the transcription- and actin-related functions of JMY in cellular processes, including early embryo development. Resolving these questions will contribute to our understanding of the cellular function of JMY and the general mechanisms that regulate actin dynamics in the cytosol and nucleus.
The developmental block of JMY-deficient embryos might coincide with the onset of zygotic genome activation (ZGA). ZGA is an essential step for maternal-to-zygotic-transition and results in a novel gene expression, which establishes the totipotent state of each blastomere at the cleavage-stage embryo (Hai et al., 2014). The first endogenous transcription occurs in the late zygotic stage in mice (Niu et al., 2014), and inhibition of RNA polymerase II with α-amanitin results in a block at the 2-cell stage (Fujii & Naito, 2013). Therefore, we tested the ability of JMY-deficient mouse embryos to activate the zygotic genome by performing RT-PCR of several genes that have been recently reported to be markers of ZGA (Bultman et al., 2006; Niu et al., 2014). The mRNA expression of the eukaryotic translation initiation factor 1A (eIF-1α), importin α5, heat shock protein 70.1 (hsp70.1), and murine endogenous retrovirus-like gene (MuERV-L) was analyzed in early JMY-deficient embryos. In addition, the transcript of the ZGA marker gene, metallothionein 1A (MT1A), was also detected in this study. In addition to its roles in cell mobility (Bultman et al., 2006), JMY can also function as a transcriptional coactivator (Zeng & Schultz, 2005).
In summary, CRISPR/Cas-mediated genome editing represents an efficient and a simple method for generating genetic modifications in mice in one step. JMY is involved in the early embryonic development in mouse before implantation, and acts as a transcription cofactor and as a ZGA-related gene.