
INTRODUCTION
Lung cancer is the major common cause of cancer-related mortality worldwide. In 2018, there were over 1.76 million deaths globally due to lung cancer, amounting to about 18.4% of all cancer-related deaths, and new lung cancer cases account for 11.6% of all new cancer cases (WHO, 2020). Tobacco smoking is the most significant risk factor for development of lung cancer. Compared with never-smokers, those who smoke tobacco have about 20-fold higher relative risk of developing lung cancer. Most lung cancers are known as carcinomas, and non-small cell lung cancer (NSCLC) comprises of approximately 85% of all lung cancers, with about 60% diagnosed at advanced metastatic stage when therapeutic option is no longer available (Planchard et al., 2018).
Most cancer cells, including NSCLC, exhibit diverse genetic alterations, which define as driver mutations contributing to the molecular pathogenesis of cancer. Over 60% of NSCLC harbor various oncogenic driver mutations, and these mutations in membrane receptors and downstream effector proteins lead to uncontrolled cell growth, proliferation and survival via a complex cascade of cell signaling pathways such as RAS/RAF/MEK/ERK or PI3K/AKT/mTOR pathway (Chan & Hughes, 2015). Activating mutations in epidermal growth factor receptor (EGFR), anaplastic lymphoma kinas (ALK), v-ros avian UR2 sarcoma virus oncogene homolog 1 (ROS1), Kirsten rat sarcoma viral oncogene homolog (KRAS), and v-RAF murine sarcoma viral oncogene homolog B (BRAF) have been found as oncogenic driver mutations in NSCLC (Ding et al., 2008; Govindan et al., 2012; Collisson et al., 2014). Thereafter, significant progress has been made to develop molecularly targeted agents for more personalized treatment of NSCLC patients, and several small molecule inhibitors, such as erlotinib and gefitinib targeting mutated EGFR, have been developed and used as monotherapy or in combination with chemotherapy (Schrank et al., 2018).
Metformin belongs to biguanide derivative that has been widely used as a first-line medication for type II diabetes mellitus for over 60 years and now prescribed to about 120 million diabetic patients worldwide. Metformin decreases blood glucose levels by inhibiting gluconeogenesis in liver and increasing uptake and utilization of glucose by skeletal muscle (Shaw et al., 2005). A retrospective study by Evans et al. (2005) suggested for the first time that metformin treatment is associated with the reduced risk of all cancers compared with other antidiabetic treatments in diabetic patients. Following studies showed that diabetic patients using metformin decrease lung cancer risk at the magnitude of 39%–45%, compared with those not using metformin (Hall et al., 2005; Lai et al., 2012). Several in vitro or in vivo preclinical studies also revealed diverse anticancer effects, in which metformin treatment results in a significant decrease in cell proliferation, tumor growth and colony formation, and induces apoptosis and cell cycle arrest in various human lung cancer cell lines (Ashinuma et al., 2012). In addition to monotherapy, combination of metformin with other chemotherapeutic or molecular targeted agents was shown to potentiate synergistically the antitumor effect (Morgillo et al., 2013; Tseng et al., 2013). Moreover, clinical trial also showed promising results, in which metformin treatment in combination with gemcitabine/cisplatin in non-diabetic and metastatic NSCLC patients significantly improves the objective response rate, overall survival and media progression free survival without significant increase in toxicity (Sayed et al., 2015).
The molecular mechanisms for the antitumor effect of metformin have been suggested but revealed as a much more complex nature (Vancura et al., 2018). The most well-known effect of metformin is the inhibition of complex I in the mitochondrial electron transport chain, which leads to increasing the intracellular AMP/ATP ratio. The high AMP/ATP ratio in turn phosphorylates and activates adenosine monophosphate activated protein kinase (AMPK), a heterotrimeric serine/ threonine protein kinase which regulates the multiple signaling pathways involved in cancer cell proliferation, including the suppression of PI3K/AKT/mTOR pathway (Griss et al., 2015). Metformin-mediated AMPK activation and mTOR inhibition suppress cell proliferation through reducing phosphorylation of its major downstream targets, the 70 kDa ribosomal protein kinase S6 (p70S6K) and eukaryotic initiation factor 4E-binding protein1 (4E-BP1) (Shaw et al., 2005). In contrast to metformin-induced inhibition of PI3K/AKT/mTOR pathway, there is contradictory effect of metformin on RAS/RAF/MEK/ERK pathway in NSCLC cells. Several studies showed that metformin inhibited ERK activation (Do et al., 2013; Ko et al., 2019), while activation of ERK in response to metformin was also reported (Morgillo et al., 2013). Considering the presence of compensatory loops that activate one pathway following the blockade of the other signaling cascade especially in cancer cells with RAS mutation (De Luca et al., 2012), the activation of ERK could result from inhibition of PI3K/AKT/mTOR pathway in response to metformin treatment, requiring blockade of both pathways for more efficient antitumor effect.
The present study, therefore, undertook to determine the combined effect of metformin and trametinib, a MEK inhibitor, on cell viability in NSCLC cell line NCI-H2087 with coexistent mutations of BRAF and NRAS. Here, we show that metformin induces the activation of ERK, and the combination of metformin and trametinib gives synergistic effect on cell survival in treatment with low doses, and antagonistic effect when treated two drugs with high doses.
MATERIALS AND METHODS
The human NSCLC cell line NCI-H2087 was purchased from Korean Cell Line Bank (Seoul, Korea). The cells were cultured in RPMI 1640 (Sigma-Aldrich, Gillingham, UK) supplemented with 10% (vol/vol) heat inactivated fetal bovine serum (Gibco BRL, Grand Island, NY, USA) and 1% streptomycin/penicillin at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were maintained mycoplasma free by treating 5 μg/mL of Plasmocin (InvivoGen, California, CA, USA). Trametinib was obtained from LC Laboratories. The compound was initially dissolved in dimethyl sulfoxide (DMSO, Sigma-Aldrich) to a concentration of 1 mM and further diluted in RPMI 1640 media. Metformin (also known as 1,1-dimethylbiguanide hydrochloride) was purchased from Sigma-Aldrich and dissolved in RPMI 1640 media to a working concentration of 100 mM.
MTT assay was applied to measure cell viability as described previously (Kim et al., 2018). Briefly, cells were harvested and seeded in 24-well plates at a concentration of 5×104 cells/well for 24 h. Then, cells were treated with increasing concentrations of trametinib (2.5–40 nM), metformin (0.25–4 mM), their combinations or vehicle control for 72 h. Experiments were performed in triplicate, each conducted in quadruplicate. The IC50 values (concentrations of drugs resulting in 50% decrease in cell viability relative to controls), combination index (CI) and drug reduction index (DRI) were calculated using CompuSyn software (ComboSyn). The CI value is a quantitative measure of the degree of drugs interaction. According to the recommendation of Chou-Talalay (Chou & Talalay, 1981), CI<1 indicates synergistic effects of drugs; CI=1 indicates additive effect; CI>1 indicates antagonism. DRI denotes how many folds of dose reduction are allowed for each drug due to synergism as compared to the dose of each drug alone.
Western blotting assays were carried out as previously described (Kim et al., 2018). Primary antibodies included pERK1/2 (Tyr204), ERK1/2, cyclin D1, β-actin (all from Santa Cruz Biotechnology, Dallas, TX, USA), and p4E-BP1 (Ser65), 4E-BP1, pS6 (Ser240/244), pS6 (Ser235/236), S6, pAMPKα (Tyr172), AMPKα1/2 (all from Cell Signaling). Following incubation with secondary antibodies conjugated to horseradish peroxidase (Cell Signaling), immunoreactivity was detected with enhanced chemiluminescence method (Santa Cruz Biotechnology).
Cells were plated in 6-well culture dishes at a density of 600 cells per well. After 24 h, cells were treated with metformin, trametinib and their combination. Every three days, medium was changed with fresh medium containing the corresponding concentration of the drugs. Following 15-day treatment, cell colonies were washed with cold PBS and then fixed with ice-cold 100% methanol. Cells were stained with 0.1% crystal violet in 20% methanol for 10 min and pictures were taken with a digital camera (Olympus).
RESULTS
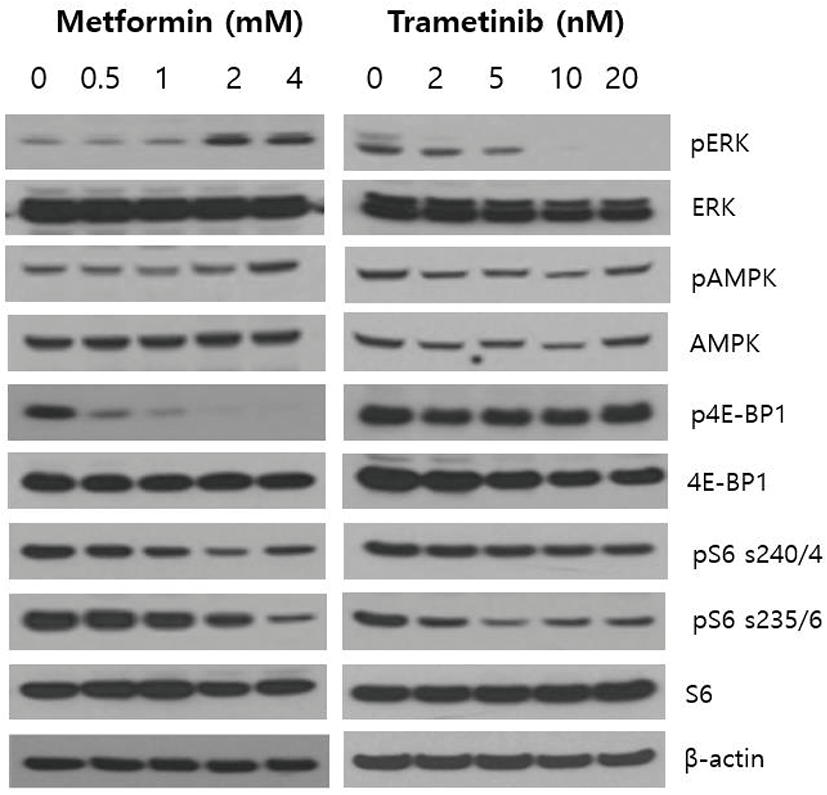
To delineate whether single agent metformin and trametinib could affect the activity of cell signaling pathways in NCI-H2087 NSCLC cells, we first analyzed the alterations of main downstream effector proteins of PI3K/AKT/mTOR and RAS/RAF/MEK/ERK pathways following treatment of metformin and trametinib for 24 h. As shown in Fig. 1, metformin suppressed the levels of p4E-BP1, pS6 (Ser235/236), pS6 (Ser240/244) in a dose-dependent manner, and increased the phosphorylation of AMPK. Interestingly, metformin increased the levels of pERK especially at high doses of 2 mM and 4 mM. On the other hand, trametinib inhibited the levels of pERK, pS6 (Ser235/236), but did not significantly affect the activity of AMPK, 4E-BP1 and pS6 (Ser240/244). These findings suggest that metformin exerts its effect in the direction of inhibiting PI3K/AKT/mTOR pathway, but simultaneously plays its effect toward activation of RAS/RAF/MEK/ERK pathway in NCI-H2087 NSCLC cells.
Next, we asked what could be happened in cell survival when combined metformin with trametinib. To this end, NCI-H2087 cells were exposed to varying concentrations of drugs as single agent or their combination for 72 h, and cell survival analysis was performed using MTT assay. As expected, relative cell viability was decreased following treatment of metformin and trametinib in a dose-dependent manner. IC50 values (concentrations of drugs leading to 50% decrease in cell viability relative to controls) for metformin and trametinib were 3.2 mM and 108.4 nM, respectively. Combination of two drugs at low doses resulted in a greater inhibition of cell viability than those of metformin or trametinib alone, while the combination treatment at high doses did not show the additively increased inhibition of cell viability (Fig. 2A). To quantify the response of NSCLC cells to the combination of metformin and trametinib, we combined two drugs in a constant ratio to each other and measured CI and DRI using CompuSyn software. The CI values in combination at low doses ranged from 0.62 (at the combination of 0.25 mM metformin and 2.5 nM trametinib) to 0.84 (at the combination of 1 mM metformin and 10 nM trametinib) indicating synergism according to the method of Chou-Talalay (Chou & Talalay, 1981). On the other hand, CI values in combination at high doses were 1.33 (at 2 mM metformin and 20 nM trametinib) and 2.08 (at 4 mM metformin and 40 nM trametinib) denoting antagonistic effect between two drugs (Fig. 2B). The DRI values displayed a similar pattern to that of CI values; combination of two drugs at low doses showed a remarkable drug reduction effect with DRI Values above 1, and at high doses little drug reduction effect with DRI values of metformin below 1 (Fig. 2C).
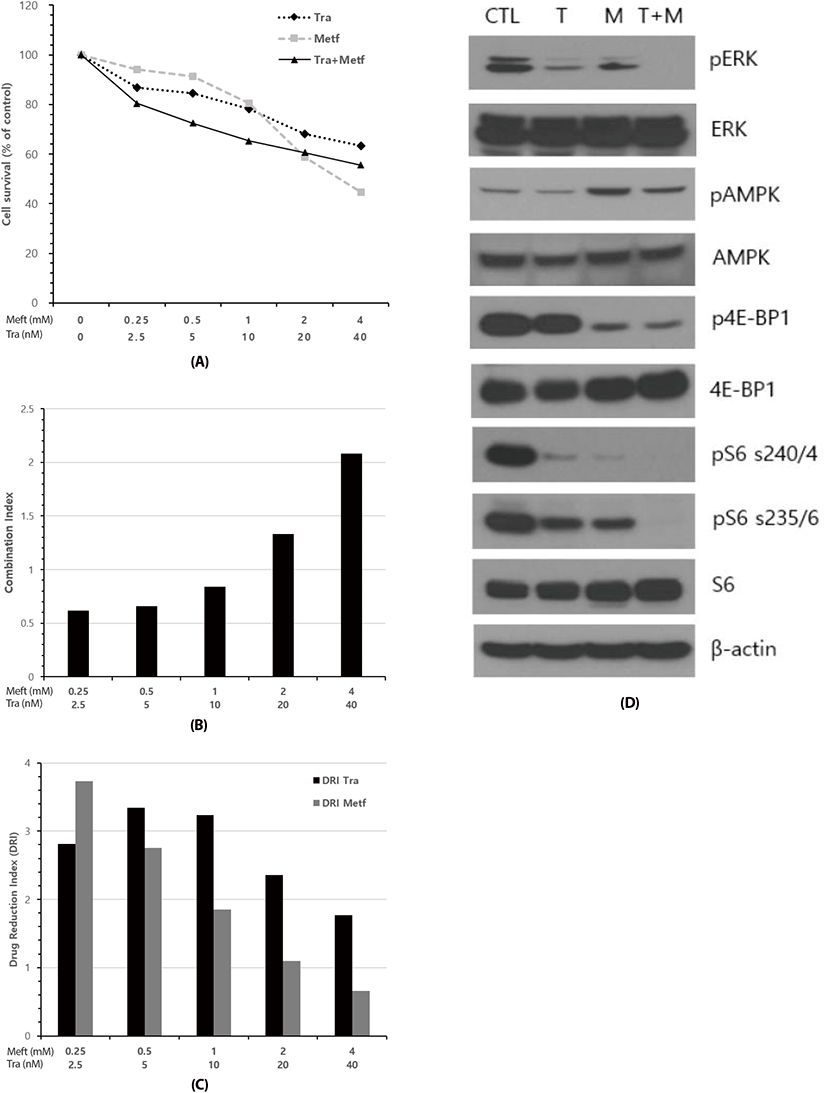
To evaluate the mechanisms underlying the synergistic growth inhibitory effect between metformin and trametinib at low doses, we characterized the effect of this drug combination on downstream regulatory proteins of cell signaling pathways involved in cell survival and proliferation using Western blot analysis. After 24 h of treatment, the combination of metformin and trametinib led to the enhanced suppression of pERK, p4E-BP1, pS6 (s240/244), pS6 (s235/236) compared to that of single agent treatment, and trametinib revealed little effect on metformin-induced activation of AMPK (Fig. 2D).
In the present study, we showed that the combination of metformin and trametinib in treatment with low doses synergistically inhibits NSCLC cancer cells. To extend these results into long-term effect of the combination treatment, we tried colony formation assay by culturing NCI-H2087 NSCLC cells for 15 days. As shown in Fig. 3, treatment with metformin and trametinib alone resulted in a partial inhibition of colony formation, whereas the combination treatment with low doses enhanced the inhibitory effect on the formation and growth of cell colonies as compared with either agent alone. These results further support the synergistic growth inhibitory effect of metformin and trametinib combination on cell viability assayed by using MTT assay.

DISCUSSION
Conventional clinical therapeutics for NSCLC depends on surgical resection of tumor mass, radiation therapy and chemotherapy. Chemotherapeutic strategies, such as cisplatin monotherapy or platinum-based combination therapy, give limited benefits due to drug resistance and serious side effects resulting from killing of normal cells (Stinchcombe et al., 2010). In NSCLC, diverse molecular alterations in oncogenic genes involved in cell survival and proliferation have been known, such as EGFR, RAS, PI3k, and BRAF. Recently, several small molecule inhibitors targeting these oncogenic mutations have been developed and proved their efficacies in the treatment of NSCLC. Although these targeted agents achieved remarkable progress for NSCLC patients with better overall response rate and progression-free survival compared with chemotherapeutic agents, most patients eventually experience relapse due to acquired drug resistance to these agents (Liu et al., 2020). Therefore, the new therapeutic strategies to overcome the acquired resistance is required in the field of NSCLC oncology and current research is focusing on multidrug combination therapy with minimal side effects.
The antidiabetic drug metformin proved to be well-tolerable drug with safety profile and low cost. Moreover, there is no report for acquired resistance to metformin although it has been used as first-line treatment for type II diabetes over 60 years. Metformin as monotherapy or combination therapy inhibits the growth and proliferation of NSCLC cell in vitro and in xenograft model (Yousef & Tsiani, 2017). Our previous studies also suggested that metformin inhibits mTOR activity via activation of AMPK and suppresses the phosphorylation of mTOR substrates, 4E-BP1 and S6 in melanoma and colorectal cancer cells. We also showed that metformin inhibits the activation of ERK, a key mediator of RAS/RAF/MEK/ERK proliferative signaling pathway (Kim et al., 2018; Ko et al., 2019).
However, the effect of metformin on RAS/RAF/MEK/ERK pathway has shown some controversial results in NSCLC cells. Several studies suggested that metformin treatment inhibits ERK phosphorylation in diverse NSCLC cells (Do et al., 2013; Ko et al., 2013). Morgillo and his collaborators (2013), on the other hand, showed that exposure of metformin to NSCLC cells leads to phosphorylation and activation of ERK through an increased BRAF/CRAF heterodimerization. Our present study also showed a remarkable increase of ERK phosphorylation and suppression of mTOR signaling pathway following metformin treatment in NCI-H2087 NSCLC cells. Although crosstalk and compensation effect between signaling pathways have been well recognized (De Luca et al., 2012), the molecular mechanism or cellular context underlying metformin-induced ERK activation is unclear. Since BRAF/CRAF heterodimerization is a key event for the activation of ERK specifically in RAS mutant cells (Holderfield et al., 2014), metformin-induced activation of ERK is most probable in cells with activated RAS. However, activation of ERK after metformin exposure is observed in NSCLC cells with wild-type RAS (Corte et al., 2016). In addition, Martin and his colleagues (2012), using melanoma cell with BRAF mutation, demonstrated that metformin-induced AMPK activation targets and reduce the DUSP6 protein, a phosphatase acting as ERK-negative regulator, which results in increased ERK activity and acceleration of cell growth.
The metformin-induced activation of ERK could be important in cancer therapeutics, since metformin, via activation of ERK, could compromise its well-known anticancer activity suppressing mTOR signaling pathway. Therefore, we reasoned that suppression or activation of ERK activity in response to metformin treatment could be the predictive biomarker for synergism or antagonism when combined metformin with other molecular targeted agents. To elucidate this subject, we combined metformin with trametinib, a MEK inhibitor, and found that the combination synergistically decreases cell viability in treatment with low doses of two drugs, while it gives antagonistic effect at high doses. Our result is inconsistent with others (Corte et al., 2016) in which the combination of metformin with MEK inhibitor selumetinib induces synergistic anti-proliferative effect in NSCLC cell lines, independently from the RAS mutational status. In the present time, we have no idea for this difference, but we speculate that in our cell model metformin-induced activation of ERK in combination of two drugs at low doses is not strong enough to overcome the trametinib-induced inhibition of ERK activation, but at high doses metformin counteracts the trametinib-induced inhibition of ERK activity, which in turn leads to cell proliferation and antagonistic effect. Another plausible explanation for the discrepancy may be the difference of mutational status in NSCLC cells between the studies; whereas we used NCI-H2087 cells with BRAF mutation, Corte and his colleagues employed NSCLC cells with wild type BRAF. In melanoma cells with mutant BRAF, metformin-induced increase of pERK is dependent on AMPK-induced degradation of DUSP6 protein (Martin et al., 2012). In NCI-H2087 cells with coexistent mutations of NRAS and BRAF, therefore, AMPK-mediated DUSP6 protein degradation could potentiate the pERK increase induced by BRAF-CRAF heterodimerization occurred in the context of RAS mutated cells. In BRAF wild-type cells, in contrast, trametinib may counteract pERK increase by metformin-mediated BRAF-CRAF heterodimerization, since MEK inhibitor acts downstream of RAS/RAF/MEK/ERK signaling pathway.
To our knowledge, this study is the first report to show biphasic antitumor effect in the combination therapy of metformin and trametinib using NSCLC cells. These findings suggest that the efficacy of metformin and trametinib combination therapy may depend on the alteration of ERK activity induced by metformin and specific cellular context of cancer cells such as mutational status. Further studies are required to elucidate the effect of metformin and trametinib combination in treatment with high doses, and in vivo effect of this combination therapy.