
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common cancers and the fourth leading cause of cancer-associated death (Bray et al., 2018). HCC rapidly progresses, and it is diagonized mostly at advanced stages. Thus, there are very few options for the prognosis and treatment of HCC. It has been suggested that metabolic reprogramming is a unique feature of cancer cells, including HCC. Aerobic glycolysis, known as the Warburg effect, is one of the metabolic reprogramming processes in cancer cells (Ward & Thompson, 2012). Under aerobic glycolysis conditions, cancer cells acquire an excessive amount of glucose to overcome their lower efficiency in producing enough energy molecules (ATP) for the proliferation and survival of cancer cells. Thus, the use of glycolysis-blocking agent, such as 2-deoxyglucose (2-DG) has been tested in some cancer cells but has been unsuccessful due to its toxicity and insufficient suppression of cancer cell growth at tolerable doses (Raez et al., 2013). HCC cells are different from normal hepatocytes in glucose metabolism and express different metabolic enzymes (Hay, 2016). Among several metabolic enzymes, hexokinase-2 (HK-2) is predominantly expressed in HCC cells. Because HK-2 is not expressed in normal hepatocytes but exclusively expressed in HCC cells, selective blockade of HK-2 can be a clinical target for the prevention or treatment of HCC. A recent study demonstrated that HK-2 depletion successfully inhibits HCC incidence in a mouse model (DeWaal et al., 2018).
Metformin is a widely used oral hypoglycemic drug for the treatment of type 2 diabetes (Rena et al., 2017). It is considered as a first-line therapy due to its effectiveness, safety, and low cost. Metformin works by decreasing glucose production in the liver, increasing insulin sensitivity in peripheral tissues, and reducing glucose absorption in the gut (Viollet & Foretz, 2013). Its mechanisms of action are complex and involve the activation of adenosine monophosphate-activated protein kinase (AMPK), which leads to the inhibition of hepatic gluconeogenesis and the stimulation of glucose uptake in muscle cells (Kim et al., 2008). Beyond its glucose-lowering effects, there is growing evidence that metformin has other biological effects that may provide additional health benefits. These include its anti-cancer (Zhang et al., 2013a), anti-inflammatory (Zhang et al., 2021), and anti-aging (Barzilai et al., 2016) effects. Metformin also modulates the gut microbiota (Forslund et al., 2015). Several large-scale observational studies and meta-analyses have suggested that metformin use is associated with a reduced incidence and mortality of various cancers, including breast, colorectal, pancreatic, and prostate cancer (Zhang et al., 2013b).
We previously showed that metformin induces the arrest of cell cycle progression and apoptosis in H4IIE rat HCC cells (Park, 2015). Interestingly, metformin induces a marked increase in glucose consumption, lactate output, and reactive oxygen species (ROS) generation in H4IIE cells (Park, 2019). We have also shown the anti-cancer activity of metformin in different cancer cell types (Lee et al., 2017; Ko et al., 2020). Herein, we report a new finding that metformin increases intracellular fat deposition together with the incidence of apoptosis of H4IIE cells. Since the effect of metformin on hepatic de novo lipogenesis (DNL) in HCC, not in normal hepatocytes, is not clearly understood yet, our present report is informative in understanding the metabolic diversity of HCC cells in lipogenic processes, as well as glucose usage. It is helpful in designing metabolic approaches for the prevention and treatment of HCC.
MATERIALS AND METHODS
Cell culture reagents, except fetal bovine serum (FBS), were purchased from Sigma-Aldrich Chemical (Sigma-Aldrich, St. Louis, MO, USA). FBS was obtained from Life Technologies (Rockville, MD, USA). Polyclonal and monoclonal antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). Horseradish peroxidase-conjugated secondary antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Electrophoresis reagents were obtained from Invitrogen (Carlsbad, CA, USA). Unless otherwise specified, all other reagents were purchased from Sigma-Aldrich.
H4IIE cells were obtained from the Korean Cell Line Bank (Seoul, Korea) and maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 5.5 mM glucose and 10% FBS. At the beginning of the experiments, H4IIE cells were incubated in serum-free DMEM overnight. Cells were washed with Dulbecco’s phosphate-buffered saline (D-PBS) and further incubated in glucose free DMEM (GFM) containing different nutrients and test reagents.
Cell viability was analyzed using the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazoli-um bromide] assay as described previously (Mosmann, 1983). Briefly, H4IIE cells were incubated in D-PBS containing MTT (0.5 mg/mL) for 30 min at 37°C. The cells were washed with D-PBS, and the blue-colored formazan product was subsequently solubilized in 0.5 mL of 2-propanol for 20 min. The absorbance of the converted dye was measured at a wavelength of 570 nm.
Cells were incubated in serum-free DMEM (5.5 mmol/L glucose). After treatment, the glucose concentration in the medium was measured using a glucose assay reagent (Asan Pharm, Seoul, Korea) based on the glucose oxidase method. The amount of glucose in the culture medium was subtracted from that of DMEM to calculate the glucose consumption (Yin et al., 2002). Lactate concentration in the medium was measured with a lactic acid assay kit from Megazyme (Wicklow, Ireland).
The generation of ROS within cells was measured using 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA; Halliwell & Whiteman, 2004). H4IIE cells were grown in 12-well culture plates and treated with reagents. After treatment, H2DCFDA (10 μmol/L) was added to each well and further incubated for 30 min. The supernatant was removed from each well, and the cell monolayers were dispersed with trypsin-EDTA solution. Each volume of cell suspension was transferred into a black 96-well plate, and the fluorescence intensity of DCF was measured at 485 nm/535 nm for excitation/emission using a multi-well fluorescence reader (Spectrafluor, Tecan, Austria). Results are represented as relative units.
After treatment, the cells were lysed in ice-cold lysis buffer (50 mM Tris-HCl, 1% nonidet P-40, 0.25% sodium deoxycholate, 150 mM NaCl, 1 mM sodium orthovanadate, 1 mM NaF, 1 mM phenylmethylsulfonyl fluoride, 1 mM aprotinin, 1 mM leupeptin, and 1 mM pepstatin A). Equal amounts (10–20 μg) of protein were separated using SDS-PAGE on 4%–12% polyacrylamide gels and transferred onto PVDF membranes. The membranes were incubated in blocking buffer (5% nonfat dry milk in Tris-buffered saline [TBS]-0.1% Tween-20 [TBS-T]) for 1 h at room temperature, after which the membranes were probed with different primary antibodies (at dilutions of 1:1,000–1:2,000). After a series of washes, the membranes were further incubated with the respective horseradish peroxidase (HRP)-conjugated secondary antibodies at a dilution of 1:10,000. The signal was detected using the enhanced chemiluminescence detection system (Intron, Seongnam, Korea).
The degree of nuclear condensation (a marker of apoptosis) was observed using a cell membrane-permeable DNA-specific fluorescent dye (bisBenzimide H33342 trihydrochloride, H33342). After treatment, the cells were incubated with H33342 (1 mg/mL) for 15 min, then observed under a fluorescent microscope (IX70, Olympus, Japan) and imaged using a digital camera (DP-70, Olympus, Japan).
Acidic intracellular vesicles, a marker of the onset of autophagy, were visualized using acridine orange (AO) staining. After treatment, the cells were washed twice with D-PBS and stained with 1 μg/mL AO in 2% acetone in D-PBS for 15 min at room temperature. The AO-stained cells were observed and imaged by a fluorescent microscope. Nile red is a fluorescence dye used to stain cytoplasmic lipid droplets. After treatment, the cells werce washed twice with D-PBS and then stained with 200 ng/mL nile red in 2% acetone for 15 min. The cells were washed again with D-PBS to remove excess stain, and the nile red-stained cells were observed and imaged by a fluorescent microscope.
The amount of cytoplasmic triglyceride was measured with an assay kit (TG-S, Asan Pharm) with a modification. Briefly, cells in 35-mm culture dishes were homogenized with 1 mL of lysis buffer (5% Triton X-100 in D-PBS) and heated for 5 min at 90°C. After cooling to room temperature, the homogenate was heated and cooled twice more. After centrifugation of the homogenates, the supernatant was used for triglyceride assay according to the kit’s manual. At the same time, protein concentrations were also measured by a standard BCA method to normalize the amount of triglyceride to the same amount of protein per sample.
RESULTS
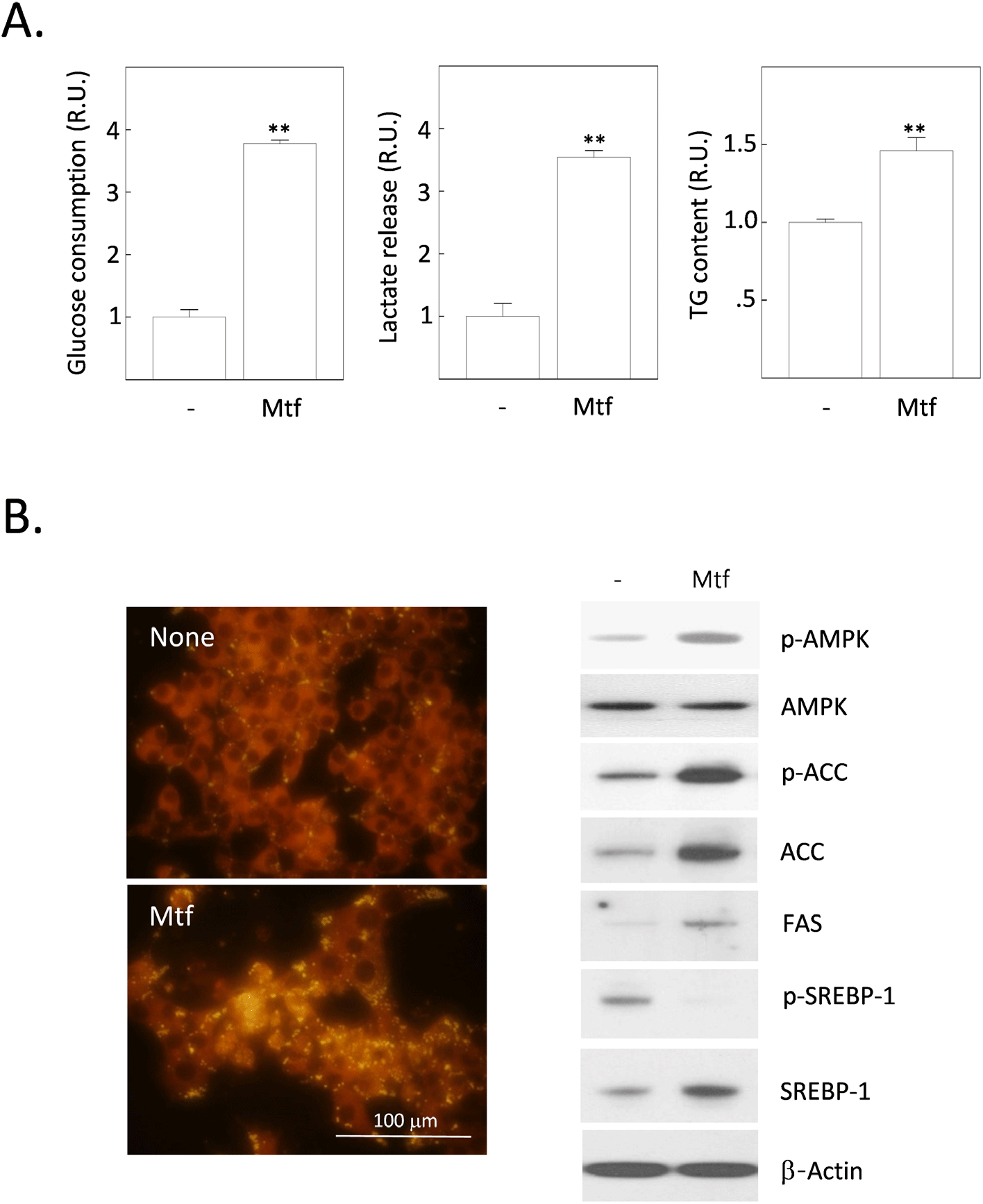
Metformin works by increasing insulin sensitivity in the liver and muscle, which helps to reduce blood sugar levels. In muscle, metformin increases glucose uptake and utilization. It also increases the rate of glycogen synthesis. In the liver, metformin inhibits gluconeogenesis, which is the process of making glucose from non-carbohydrate sources such as protein and fat. It also inhibits glycogenolysis, which is the breakdown of glycogen to glucose. Metformin also increases glycogen synthesis, which is the process of storing glucose in the liver. Additionally, metformin increases the expression of glucose transporter 2 (GLUT2), which is the main GLUT isoform in the liver. This increases the amount of glucose that can be taken up by the normal liver. However, metformin has anti-cancer properties, including the ability to inhibit the growth of HCC cells. One of the ways that metformin exerts its anti-cancer effects is by inhibiting glucose uptake in HCC cells. Glucose is a major source of energy for cancer cells, and by inhibiting glucose uptake, metformin can starve the cells and cause them to die. To verify this hypothesis, we measured the effect of metformin on the uptake (or consumption) of glucose in H4IIE HCC cells. Contrary to the hypothesis, low dose (1 mmol/L) metformin markedly increased the consumption of glucose for 24 h, approximately 3.7 times over the non-treated (control) cells (Fig. 1A). Accordingly, the release of lactate from cells into the culture medium was also increased 3.5 times over the control cells (Fig. 1A). Such increases in the glucose consumption and lactate release paralleled with a 1.5-fold increase in the cytoplasmic fat content (Fig. 1A). From the fluorescence observation after staining with Nile red after metformin treatment, the intensity of fat-specific fluorescence was also enhanced (Fig. 1B). We, therefore, examined the changes of intracellular proteins affecting the hepatic DNL processes (Fig. 1B). The phosphorylation of AMPK as well as Acetyl-CoA carboxylase (ACC) were stimulated by metformin treatment for 24 h. Expression levels of key enzymes stimulating lipogenesis including ACC, fatty acid synthase (FAS) and sterol regulatory element-binding protein 1 (SREBP1) were also increased. Interestingly, levels of phosphorylated SREBP-1 (p-SREBP1) were completely downregulated by metformin treatment.
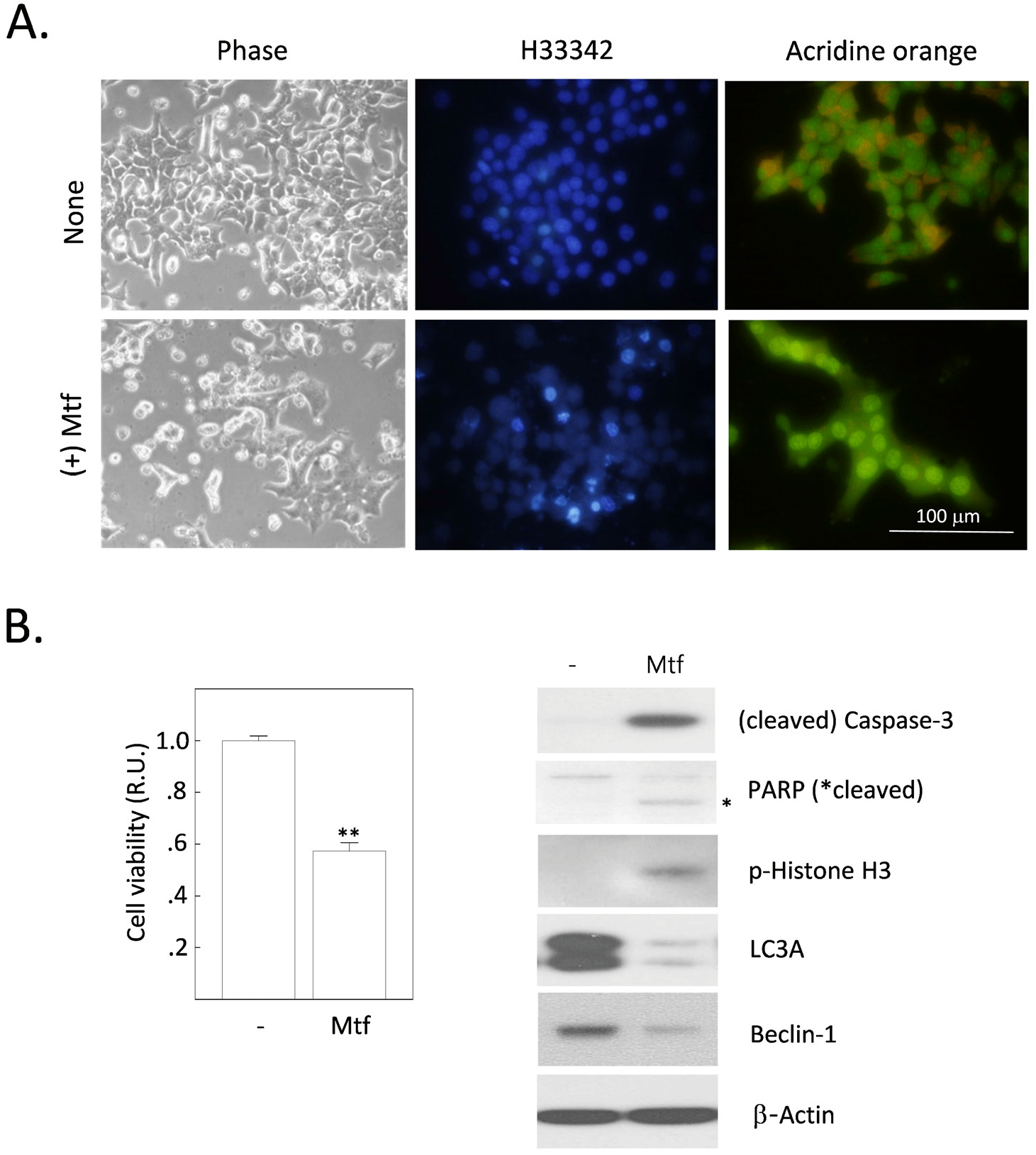
Our study showed that metformin increases intracellular fat content in H4IIE cells. We have previously reported that metformin induces apoptosis in H4IIE cells (Park, 2019). These results suggest that excessive fat deposition in cells can increase toxicity and damage (lipotoxicity or lipoapoptosis). This damage can cause a variety of health problems, especially in the normal liver, including nonalcoholic fatty liver disease (NAFLD) or type 2 diabetes. However, it is uncertain whether excessive fat deposition has the same toxic effects in HCC cells. We investigated whether metformin has any effects on cell viability, including apoptosis and autophagy. After 24 h of metformin treatment, cells were observed under a phase-contrast microscope and a fluorescence microscope after staining with H33342 or AO. The number of cells detached from culture dishes (phase-contrast) and apoptotic bodies (stained with H33342) increased with metformin treatment. However, the number of cells with autolysosomal vesicles (stained with AO) also increased with metformin treatment (Fig. 2A). The MTT assay showed that metformin decreased cell viability by 43.7% compared to untreated control cells (Fig. 2B). In addition to the morphological observations and MTT assay, we also analyzed the expression levels of several proteins associated with apoptosis and autophagy (Fig. 2B). Metformin treatment caused changes indicative of apoptosis progression (hydrolysis-induced activation of caspase-3 and PARP, phosphorylation of histone H3) and suppression of autophagy (decreased expression of beclin-1 and LC3A). In summary, metformin treatment induced autophagy and apoptosis, together with enhanced intracellular fat accumulation in H4IIE cells. These results raise further questions about the crosstalk between lipogenesis, apoptosis, and autophagy.
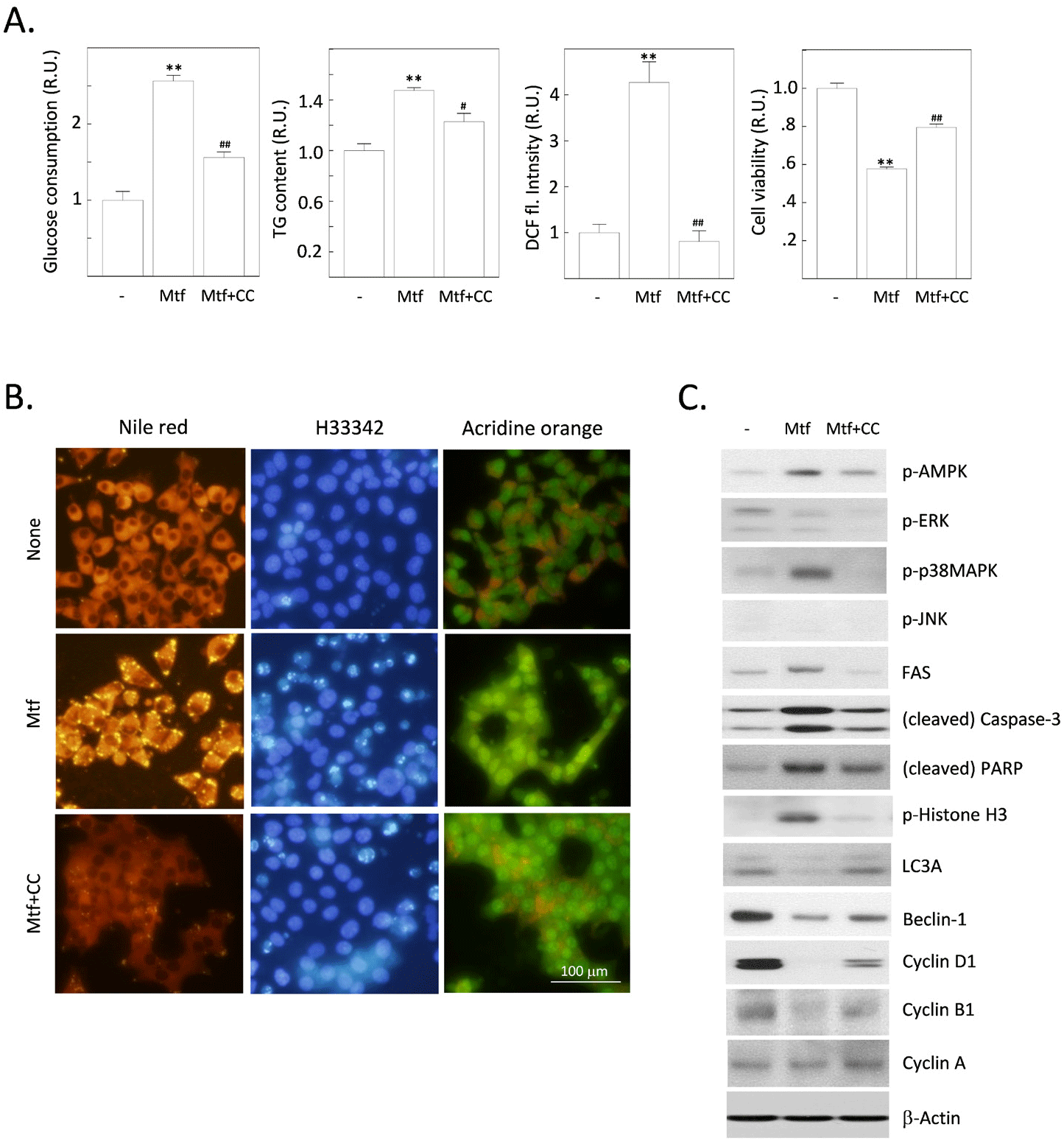
AMPK is an energy sensor that is activated when cellular energy levels are low. It is a serine/threonine kinase that phosphorylates a variety of proteins, including enzymes, transcription factors, and ion channels. It is thought to work by increasing AMPK activity. Metformin-stimulated AMPK has a number of beneficial effects in normal hepatocytes, including increased glucose uptake, decreased gluconeogenesis, increased fatty acid oxidation, reduced inflammation, improved mitochondrial function, and promoted autophagy (Wang et al., 2022). However, metformin has also been shown to have a number of other effects that are independent of AMPK activation (Rena et al., 2017). We aimed to verify whether metformin-induced changes, including the stimulation of cytoplasmic fat deposition and the progression of apoptosis, are dependent on AMPK or not. Metformin treatment stimulated glucose consumption and fat deposition and these changes are all sensitive to an AMPK inhibitor, compound C (Fig. 3A and B). Interestingly, the production of ROS was markedly stimulated by metformin, also sensitive to compound C (Fig. 3A). The formation of autolysosomal vesicles, which was reduced by metformin, was again restored by compound C. We next investigated the effect of compound C on expressions of various proteins, including signaling molecules and other proteins that regulate lipogenesis, apoptosis or autophagy, and cell cycle progression (Fig. 3C). Metformin-induced phosphorylation of AMPK was inhibited by compound C. Among MAPKs, c-Jun N-terminal kinase was not affected, and extracellular-signal regulated kinases were inactivated by metformin, but this effect was insensitive to compound C. Unexpectedly, p38MAPK was activated by metformin, but this effect was completely sensitive to compound C. Metformin-stimulated expression of FAS was suppressed by compound C. Expression of LC3A and beclin-1 was reduced metformin, and these effects were sensitive to compound C. Apoptosis-promoting changes (cleavage of caspase-3 and PARP, phosphorylation of histone H3) were induced by metformin, and these changes were all sensitive to compound C. Finally, expressions of cyclin D1 and B1, which are responsible for G1/S- and G2/M progression of cell cycle checkpoints respectively, were inhibited by metformin in compound C-sensitive manners. Collectively, metformin induces fat accumulation, onset of apoptosis, inhibition of autophagy, and cell cycle progression at G1/S and G2/M checkpoints. These events are all sensitive to the inhibition of AMPK, suggesting that AMPK mediates a series of metformin’s activity shown in our results.
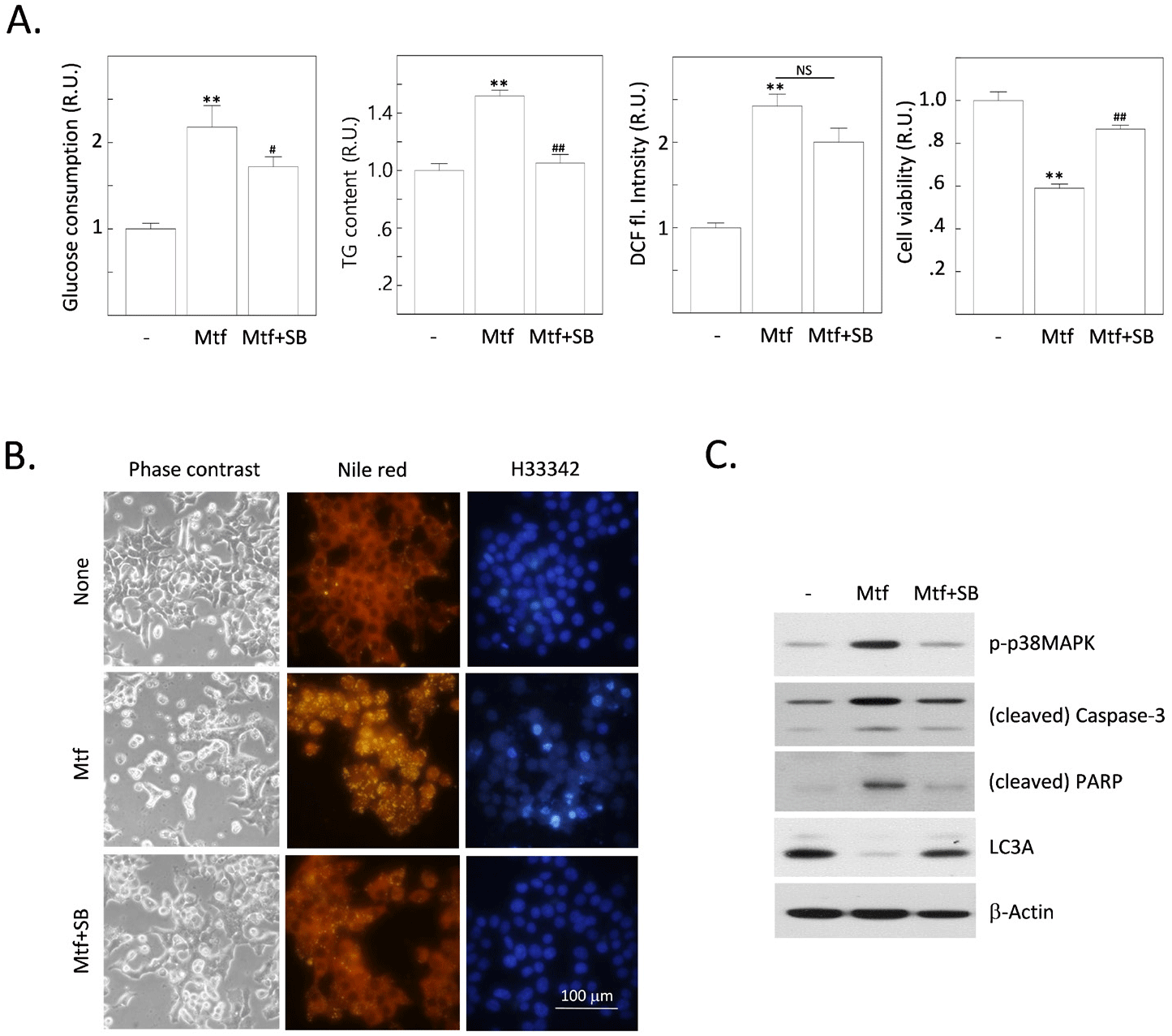
We found that metformin stimulates p38MAPK activity. This is shown by the increased phosphorylation of p38MAPK in cells treated with metformin. The inhibition of AMPK completely suppresses the metformin-stimulated p38MAPK activity. This suggests that AMPK is required for metformin-induced p38MAPK activation. To further investigate this, we tested whether the inhibition of p38MAPK can interfere with metformin-induced changes in H4IIE cells. The inhibition of p38MAPK led to the same effects as the inhibition of AMPK, including increased fat accumulation, ROS production, and the onset of apoptosis. This suggests that p38MAPK is downstream of AMPK in the metformin signaling pathway. Inhibition of p38MAPK also inhibited metformin-induced cleavage of caspase-3 and PARP, and reversed the metformin-reduced expression of LC3A. This suggests that p38MAPK is required for metformin-induced apoptosis. These results suggest that AMPK and p38MAPK are subsequently stimulated by metformin. These stepwise activations of the AMPK-to-p38MAPK pathway might play crucial roles in inducing metabolic responses (enhancement of glucose consumption and the following fat accumulation), oxidative stress by excess ROS generation, and finally the onset of apoptosis together with the reduced autophagic responses.
DISCUSSION
Metformin is a biguanide drug that is used to treat type 2 diabetes mellitus. It has been shown to have a number of beneficial effects on lipid metabolism, including reducing hepatic lipogenesis in the healthy normal hepatocytes (Stumvoll et al., 1995). Hepatic lipogenesis is the process by which the liver synthesizes triglycerides from fatty acids and glucose. Triglycerides are a type of fat that is stored in the liver and other tissues. When there is too much triglyceride production, it can lead to fatty liver disease. It also has a number of beneficial effects on lipid metabolism, including reducing hepatic lipogenesis. This can help to protect against fatty liver disease and other metabolic disorders. So far, metformin is a safe and effective drug that can be used to treat type 2 diabetes mellitus and hepatic disorders NAFLD.
A number of studies also have shown that metformin can reduce the risk of HCC in patients with chronic liver disease. For example, metformin was associated with a 40% reduction in the risk of HCC in patients with NAFLD (Zhang et al., 2013b). Another study found that metformin prevents HCC by suppressing hepatic progenitor cell activation in a rat model of cirrhosis (DePeralta et al., 2016). These studies suggest that metformin may be a useful preventive agent for HCC in patients with chronic liver disease. However, more research is needed to confirm these findings and to determine the optimal dose and duration of metformin therapy for HCC prevention.
We previously showed that metformin induces apoptosis of cultured rat HCC cells and various changes are involved in this process (Park, 2015, 2019). Metformin-induced changes include increases in the glucose consumption rate, lactate production, ROS generation, and the suppression of autophagy. Our present finding is that metformin increases the amount of fat deposited in the cytoplasm together with the generation of ROS in AMPK- and p38MAPK-dependent manners in cultured HCC cells. One of previous studies reported that metformin suppresses hepatic lipogenesis and the growth rate in HCC cells and mice liver (Bhalla et al., 2012). However, in their experiments using HCC cells, the dose of metformin and the duration of treatment is too high (up to 10 mM) and long (72 h). Moreover, AMPK is not activated in the liver of metformin-treated mice. We think that our experimental design using culture conditions with the low dose (1 mM) and minimal duration (24 h) in metformin treatment is more reasonable for investigating the effect of metformin on the lipogenesis in the cultured HCC cells. These results are different to a hypothesis suggesting the anti-lipogenic effect of metformin in the normal hepatocytes and in the liver of healthy subjects (Zhang et al., 2021). We hypothesize that the metformin-derived metabolic regulation is different or even opposite between in normal hepatocytes and HCC cells because the metabolic alterations found in the HCC cells. In the normal hepatocytes, metformin appears to reduce hepatic lipogenesis by a number of mechanisms (Han et al., 2019). First, it increases the activity of the enzyme AMPK. AMPK is a metabolic sensor that helps to regulate energy balance. When AMPK is activated, it inhibits the production of triglycerides. Second, metformin inhibits the activity of the enzyme sterol regulatory element binding protein 1c (SREBP1c). SREBP1c is a transcription factor that regulates the expression of genes involved in lipid synthesis. When SREBP1c is inhibited, it reduces the production of triglycerides. However, in our study using HCC cells, metformin stimulated the cytoplasmic accumulation of fat in a AMPK-dependent manner. Additionally, metformin upregulated the expression of FAS, ACC, and SREBP1, key enzymes responsible for the induction of lipogenesis. Thus, the effect of metformin on the lipogenesis should be addressed distinctly in the malignant HCC cells. In fact, metabolic alterations known as the Warburg effect, have been accepted as one of typical epigenetic alterations observed in cancer cells (Vaupel et al., 2019). First, cancer cells rely on glycolysis, the breakdown of glucose, for energy production. This is even in the presence of oxygen, a phenomenon known as the Warburg effect. As a result, cancer cells produce more lactate than normal cells. Lactate is a byproduct of glycolysis. Second, cancer cells take up more glucose and amino acids than normal cells. This is because glucose is the main source of energy for glycolysis and amino acids are used to build proteins, which are essential for cell growth and division. Third, cancer cells use glucose less efficiently than normal cells. This is because they have decreased mitochondrial function including oxidative phosphorylation. Fourth, cancer cells make more fatty acids than normal cells. Fatty acids are used to build cell membranes and other cellular components. However, little is known about the effect of excess cytoplasmic fatty acids on cancer cells. Lipotoxicity is a condition that occurs when cells are exposed to too much fat. This can damage the cells and lead to cell death. Lipotoxicity is a common problem in cancer cells. Cancer cells have a high demand for energy, and they often use fatty acids as a source of energy. When cancer cells take up too much fat, it can damage the cells and lead to cell death. Thus, we hypothesize that metformin-induced ‘excess’ glucose consumption can upregulate and then can produce too much ROS and following oxidative stress DNL in H4IIE HCC cells. This lipotoxicity can also lead to the death of cancer cells by a process called apoptosis. Apoptosis is a programmed cell death. When cancer cells are exposed to too much fat, they can undergo apoptosis. This can help to stop the growth of the tumor. A recent study also suggests that pharmacologically induced lipotoxicity represents a promising therapeutic strategy for the treatment of hepatocellular carcinoma (Rudalska et al., 2021). Therefore, lipotoxicity is a promising new target for cancer therapy. By targeting lipotoxicity, researchers may be able to develop new drugs that can kill cancer cells and stop the growth of tumors. Our finding provides evidences to support that notion because metformin evidently induces intracellular DNL, oxidative stress, and apoptosis in HCC cells.
The role of AMPK in lipotoxicity has been tested in non-cancerous subjects (Krishnan et al., 2023). They found that AMPK activation protected against the development of NAFLD in mice. Other study also suggests that AMPK activation increased the expression of genes involved in fatty acid oxidation and decreased the expression of genes involved in DNL (Hardie & Pan, 2002). AMPK activation has been shown to induce apoptosis in a variety of cancer cells (Kim et al., 2016). There are a number of ways that AMPK can induce apoptosis in HCC cells (Ma et al., 2020). One way is by activating the p53 tumor suppressor gene. p53 is a transcription factor that controls a number of genes involved in cell cycle arrest, apoptosis, and DNA repair. The long-term activation of AMPK leads to accelerated p53-dependent cellular senescence (Jones et al., 2005). Another way that AMPK can induce apoptosis in HCC cells is by inhibiting the mTOR pathway. mTOR is a kinase that promotes cell growth and proliferation. AMPK activation inhibits mTOR, which leads to cell cycle arrest and apoptosis (Schuster et al., 2015). Finally, AMPK can also induce apoptosis in HCC cells by promoting autophagy. Autophagy is a process by which cells degrade damaged or unnecessary proteins and organelles. AMPK activation stimulates autophagy, which can lead to the death of HCC cells (Wang et al., 2022). Overall, AMPK is a promising target for the treatment of HCC. AMPK activation can induce apoptosis in HCC cells through a number of different mechanisms. Further research is needed to develop AMPK-targeted therapies for HCC.
One of the pathways involved in lipotoxicity is the p38 MAPK pathway. p38 MAPK is a protein kinase that plays a role in cell signaling. When activated, p38 MAPK can lead to the production of inflammatory proteins and the death of cells. A recent study showed a link between lipotoxicity and p38 MAPK activation (Yang et al., 2022). p38MAPK inhibitors also can be used to treat cancer (Yong et al., 2009). Other study showed that hepatic activation of p38MAPK induces severe liver steatosis (Liu et al., 2022). In HCC cells, oxidative stress induces activation of p38MAPK (Sun et al., 2022). The findings of these studies suggest that p38 MAPK may be a target for the treatment of lipotoxicity. Our data also showed that metformin stimulates ROS generation and p38MAPK activity, which are sensitive to AMPK inhibition. One interesting result is that metformin interferes autophagy in H4IIE cells in a AMPK-dependent manner. It is still controversial whether the progression of autophagy-related events is the sign for the survival or the death of cancer cells (Das et al., 2021). In our study, autophagy may act as a survival factor because the inhibition of AMPK together with metformin treatment promotes the survival and the progression steps of autophagy in H4IIE HCC cells.
Taken together, our findings suggest that metformin induces excess glucose-induced DNL, oxidative stress by ROS generation, subsequent activation of p38MAPK, suppression of autophagy, and ultimately apoptosis.