
INTRODUCTION
The uterine epithelium is the most dynamic changing tissue according to the estrous cycle. Especially, the luminal and glandular epithelium of the uterus undergo cellular proliferation and regression. The process is precisely and rapidly regulated by hormonal regulation such as estrogen (E2) and progesterone (P4), which is very important for preparation of successful embryo implantation in the uterus. However, the regulatory networks of cellular regression in the uterus were poorly understood.
Autophagy is a cellular mechanism removing unnecessary or worn-out materials by capturing and sealing of them within a double-membrane vesicle called an autophagosome. The outer membrane of the autophagosome fuses with the lysosomal membrane to form an autolysosomal lysosome. Following fusion, the monomembrane vesicles are released into the lysosomal lumen and are degraded with the other cargo by autolysosomal hydrolysis. The covalent lipidation of microtubule-associated protein 1 light chain 3 (LC3)—a member of the ATG8 family of proteins—is a major regulatory pathway that promotes the induction of autophagy (Dikic & Elazar, 2018). LC3 is cleaved to liberate the c-terminal glycine that is necessary for ligation to phospholipids in autophagy. The lipidated form of LC3 (LC3-II) is localized to the autophagosomal membrane and is subsequently degraded upon the formation of autolysosomes (Pankiv et al., 2007; Stolz et al., 2014; Zaffagnini et al., 2018). The LC3-II is localized to the autophagosomal membrane and is subsequently degraded upon the formation of autolysosomes. Therefore, LC3 is an essential component of the autophagy machinery and plays a central role in autophagosome formation and subsequent autophagosome-lysosome fusion factor for autophagy including autophagosome formation and autolysosome formation (Nakatogawa et al., 2009; Mizushima et al., 2010). Despite this functional importance, the regulatory mechanisms of LC3 activity and its role in autolysosome formation in the uterine endometrium remain poorly understood.
Recent studies have reported that STK4 (known as a major kinase in hippo signaling pathway) was involved in autophagy regulation by phosphorylation of threonine at position 50 (T50) of LC3 protein. The study reported that LC3 phosphorylation by STK4 is important for the cellular elimination of intracellular bacteria, an established cargo for fusion and autophagy by autophagosomes and lysosomes (Wilkinson & Hansen, 2015; Wilkinson et al., 2015). Interestingly, we revealed that the expression of STK4 in the endometria of mice is regulated via estrogen during the estrous cycle in the previous study (Moon et al., 2019). So far there have been no reports on relationship between LC3 activity and regulatory signaling in the uterus. Therefore, we hypothesized that LC3 activity is regulated by STK4 during known signaling in the uterus.
In this study, we investigated how estrogen regulates late-stage autophagic maturation in mouse endometrial epithelium, with particular focus on the role of STK4-mediated LC3B phosphorylation. We demonstrate that estrogen signaling regulates LC3B phosphorylation at threonine 50 via STK4 using ovariectomized (OVX) mice and endometrial cell culture systems. Importantly, this phosphorylation specifically regulates late-stage autophagic maturation, consistent with control at the autophagosome–lysosome fusion step.
In the present study, we not only created fluorescent reporter systems for LC3, but also used OVX mouse model and target gene knockdown system to study its regulatory mechanism and signaling pathway in the uterine epithelium. These findings reveal a novel stage-specific regulatory mechanism linking estrogen receptor signaling with Hippo pathway kinases in autophagy control, with potential implications for understanding endometrial physiology and pathology (Table 1).
RT-PCR, reverse transcription-polymerase chain reactions; q-PCR, quantitative polymerase chain reaction.
MATERIALS AND METHODS
Seven-week-old Institute of Cancer Research (ICR) mice were obtained from KOATECH (Pyeongtaek, Korea) and housed under standardized environmental conditions with a 12-h light/dark cycle, constant temperature, and ad libitum access to food and water, as previously described (Lee et al., 2025; Park et al., 2025).
Collected uteri were fixed in 4% paraformaldehyde for histological and immunofluorescence analysis, following previously described protocols (Jang et al., 2017; Thang et al., 2023; Hwang et al., 2025).
Primary antibodies used in this study included: anti-LC3B (ab48394, dilution ratio, 1:500; Abcam, Cambridge, UK), anti-LC3B (phospho T50) (ab204297, dilution ratio, 1:100; Abcam), anti-LAMP2 (NBP2-22217, dilution ratio, 1:500; Novus, Centennial, CO, USA), anti-STX17 [fluorescein isothiocyanate (FITC)] (orb190347, dilution ratio, 1:200; Biorbyt, Cambridge, UK), and anti-β-actin (sc-47778, dilution ratio, 1:1,000; Santa Cruz Biotechnology, Dallas, TX, USA). All antibodies were diluted in 4% bovine serum albumin in phosphate-buffered saline containing 0.05% Tween 20 (PBST).
Colocalization quantification was performed blinded to treatment groups using ImageJ/FIJI software. A minimum of 50 cells per biological replicate were analyzed (n=5 animals for in vivo; n=3 independent experiments for in vitro). LC3 and LAMP2 fluorescence signals were converted to 8-bit images and thresholded independently using identical automated thresholding parameters to generate binary masks. Double-positive puncta were identified using a logical AND operation with the Image Calculator function. Quantification was restricted to defined regions of interest (ROI), and the proportion of LC3 puncta colocalized with LAMP2 was calculated as a surrogate measure of autophagosome–lysosome interaction. This object-based colocalization analysis approach is consistent with previously established methods used to assess autophagosome–lysosome fusion and autolysosome formation (De Leo et al., 2016; Matsui et al., 2018).
To investigate the effects of estrogen on the expression of autophagy-related genes in the mouse uterus, 7-week-old ICR mice were OVX and were rested for 10 days before receiving estrogen injection. Subsequent surgery was performed in accordance with the previously described procedure (Moon et al., 2019). We placed the mouse in the isoflurane chamber to anaesthetize it. Next, we placed it in a prone position, shaved the hair off its flank and disinfected the skin with a chlorhexidine solution. Then, we made an incision in the skin, pulled out the ovarian fat pad and removed the ovaries. After all procedures were completed, we placed the animal on a heating pad and maintained ad libitum access for 10 days before hormone treatment. After confirming wound healing and normal behavior, mice received subcutaneous estrogen injections. Once the surgical wound had healed and the animal’s behavior was normal, we used it for hormone treatment.
The OVX mice were treated with hormones as described in a previous study (Jeong et al., 2015). The OVX mice were injected subcutaneously with estrogen (200 ng/mouse). Their uteri were collected at 0, 1, 3, 6, 12, and 24 h after estrogen injection. To examine whether the expression of target factors in the uterus depends on estrogen, the OVX mice were either injected with estrogen (200 ng/mouse) or pretreated with the estrogen receptor antagonist ICI 182,780 (ICI) (500 μg/mouse) for 30 min before hormone treatment. Sesame oil (100 μL/mouse) was used for the control group. Each group contained five animals.
To investigate the functional role of STK4 in human endometrial epithelial cells, we employed the Ishikawa cell line as an in vitro model. Ishikawa cells were cultured in phenol red-free Dulbecco’s modified eagle medium (DMEM) supplemented with 10% charcoal-stripped fetal bovine serum (FBS) and 25% MCDB-105 medium at 37°C in a humidified atmosphere containing 5% CO2. Cells were maintained at a density of 2×103 cells/well in 24-well plates and allowed to attach for 24 h prior to transfection (Moon et al., 2019).
The cells were transfected with Stk4 small interfering RNAs (siRNAs). We used the following siRNAs: siGENOME duplex Stk4 siRNA (SR415716; Dharmacon, Lafayette, CO, USA) and universal scrambled negative control siRNA (SR30004; Dharmacon). The Ishikawa cells were transiently transfected with DNA (500 ng for each 24-well plate) or siRNA (10 μM for each 24-well plate) using Lipofectamine RNAi (Thermo Fisher Scientific, Waltham, MA, USA). Transfection efficiencies were determined by western blot analysis.
Transient transfections were performed using 1.0–2.0 μg of target DNA and Lipofectamine 2000 (Life Technologies, Waltham, MA, USA) in Opti-MEM medium, according to the manufacturer’s instructions. Ishikawa cells were transfected with expression vectors for GFP-p62, GFP-LC3, or RFP-LC3. For nuclear visualization, cells were stained with 4’,6-diamidino-2-phenylindole (DAPI) (Sigma-Aldrich, St. Louis, MO, USA) for 3 min. Autophagic puncta and protein localization were visualized using confocal microscopy.
To analyze the expression and localization of endogenous STX17 and LAMP2, cells were fixed in 1% paraformaldehyde for 30 min and subsequently washed three times with polyvinyl alcohol– phosphate-buffered saline (PVA-PBS). Nonspecific binding was blocked by incubating the cells for 1 h in a blocking buffer consisting of 5% goat serum and 0.2% Triton X-100 in PVA-PBS.
Total protein was extracted using ProNATM phospho-block solution (TransLab, Daejeon, Korea) to preserve phosphorylation status. For each sample, 30 μg of total protein was loaded onto SDS-PAGE gels and transferred onto polyvinylidene fluoride (PVDF) membranes. Western blot analysis was performed as previously described (Moon et al., 2019; Kim et al., 2025). The membranes were incubated overnight at 4°C with the following primary antibodies: anti-LC3B (ab48394; Abcam), anti-phospho-LC3B (Thr50) (ab204297; Abcam), anti-ATG5 (ABIN492606; Antibodies Online, Atlanta, GA, USA), and anti-LAMP2 (NBP2-22217; Novus Biologicals, Centennial, CO, USA). All primary antibodies were used at a dilution of 1:1,000, except for anti-β-actin (sc-47778; Santa Cruz Biotechnology), which was used at a dilution of 1:10,000 as a loading control. After incubation with appropriate HRP-conjugated secondary antibodies, the protein bands were visualized and quantified using the ChemiDoc XRS system (Bio-Rad, Hercules, CA, USA), and band intensities were normalized to β-actin.
For mammalian expression, the following plasmid constructs were used in this study: GFP–LC3B, RFP–LC3B, and GFP–p62. Cloning was performed using a TOPcloner™ Blunt Core Kit (Enzynomics, Daejeon, Korea) according to the manufacturer’s instructions. Point mutations in GFP–LC3 and RFP– LC3 were generated using the QuikChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA, USA). All primers described in Table 2. All LC3 point mutations were verified by DNA sequencing, as previously described (Wilkinson & Hansen, 2015; Wilkinson et al., 2015).
LC3, light chain 3.
Data are presented as the mean±SEM. Statistical analyses were performed using one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test for multiple comparisons. For in vivo experiments, n=5 biological replicates (animals) per group. For in vitro experiments, n=3 independent biological replicates. Technical replicates (duplicate wells) were averaged within each biological replicate before statistical analysis. A p-value<0.05 was considered statistically significant.
RESULTS
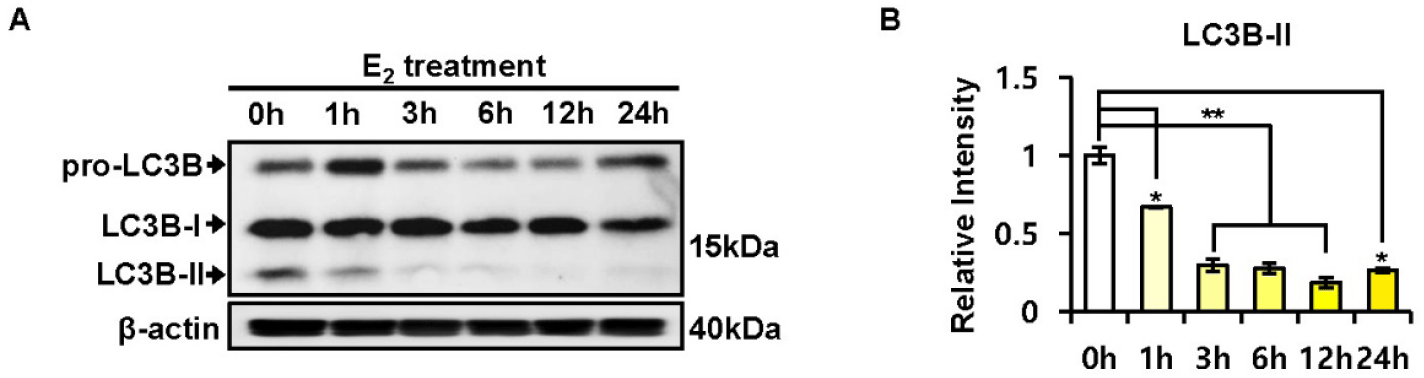
To determine whether autophagy is regulated by estrogen, the expression level of the autophagosome marker LC3B was evaluated by western blot analysis following E2 treatment in OVX mouse uteri. As shown in Fig. 1, LC3B-I levels remained unchanged (Fig. 1A), whereas LC3B-II significantly decreased from 1 h post-treatment, with minimal expression detected thereafter (Fig. 1B). These results suggest that LC3B-II dynamics are regulated by E2, consistent with late-stage autophagic maturation and increased autophagosome–lysosome fusion activity.
To determine whether E2 directly regulates late-stage autophagic maturation, we treated estrogen receptor antagonist ICI 182,780 (ICI) to OVX mice. The observation period was three hours following E2 and ICI treatments because the LC3B-II protein was clearly reduced to this timepoint at three hours of E2 treatment in Fig. 1. As shown in Fig. 2A, LC3B-II levels decreased significantly with E2 treatment, and this decrease was prevented by ICI co-treatment, indicating that estrogen receptor signaling directly regulates LC3B expression.
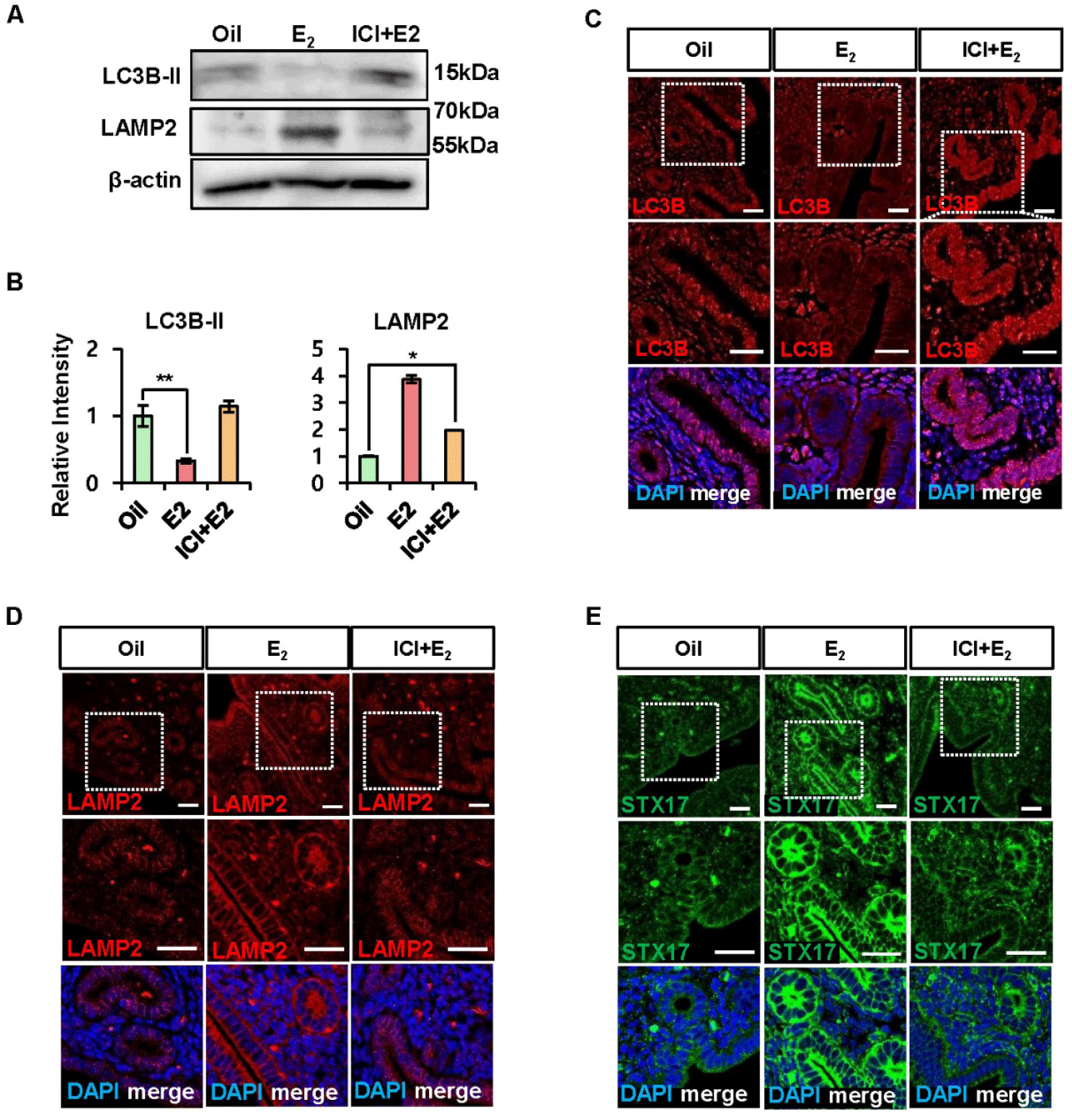
Western blot and immunofluorescence analyses were performed to examine the expression of both LAMP2, a lysosomal marker, and STX17, an autophagosome-lysosome fusion marker. LAMP2 protein levels increased after E2 treatment and were reduced by ICI pretreatment (Fig. 2A, B, and D), demonstrating that LAMP2 expression is also regulated through the estrogen receptor pathway. Immunofluorescence staining revealed that E2 treatment induced LAMP2 expression in the endometrial epithelium, which was blocked by ICI (Fig. 2D). The expression of STX17 also increased in the endometrial epithelium following E2 treatment (Fig. 2E), with a pattern consistent with that of LAMP2. These results suggest coordinate regulation of components involved in late-stage autophagic maturation (LAMP2 and STX17) by estrogen signaling, while LC3B-II decreases, consistent with enhanced autolysosomal degradation.
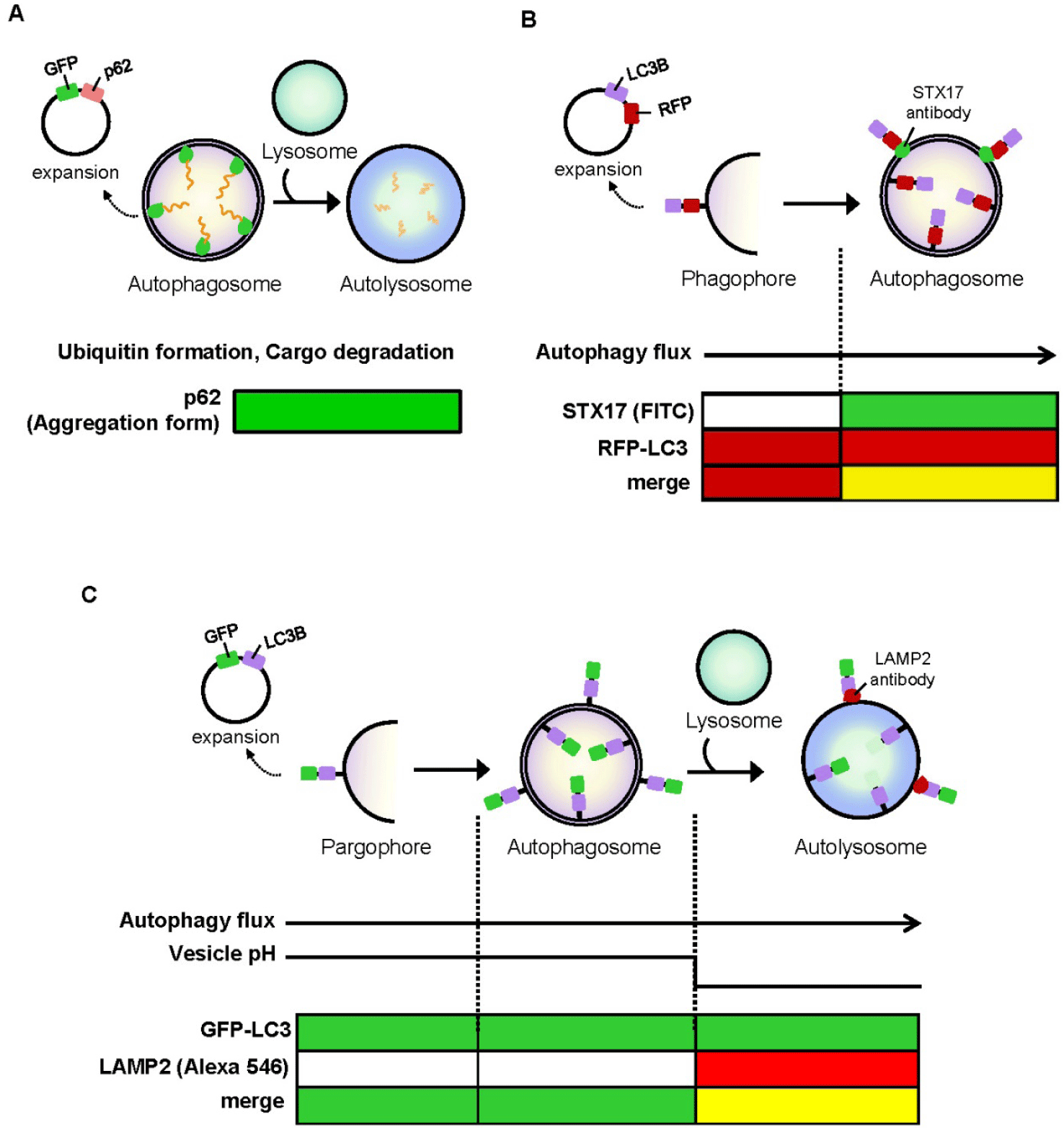
To trace the autophagic process under E2 treatment at the cellular level, we used Ishikawa endometrial cells with GFP- and RFP-tagged expression vectors. LC3B and p62 (an autophagy cargo receptor) were cloned into GFP and RFP vectors, respectively. The experimental strategy for visualizing autophagosome–lysosome fusion and autolysosomal maturation is illustrated in Fig. 3. GFP-p62 was used to monitor cargo ubiquitination and degradation during autophagosome-to-autolysosome transition (Fig. 3A). For autophagosome detection, RFP-LC3B transfection combined with STX17 immunostaining (FITC, green) produced yellow signals (merge) when autophagosome formation occurred in the cytoplasm (Fig. 3B). For autolysosome detection, GFP-LC3B transfection with LAMP2 immunostaining (Alexa 546, red) resulted in yellow signals (merge) upon autophagosome-lysosome fusion. As autophagosomes fuse with lysosomes and vesicle pH decreases, the GFP-LC3 signal is gradually attenuated within the acidic autolysosome compartment (Fig. 3C).
We next investigated whether Hippo pathway kinases STK4 regulate LC3B phosphorylation in endometrial cells. Using siRNA-mediated STK4 knockdown in Ishikawa cells, we found that phosphorylation of LC3B at threonine 50 (LC3B-T50) was significantly reduced upon STK4 depletion (Fig. 4B), indicating that STK4 directly phosphorylates LC3B at this site.
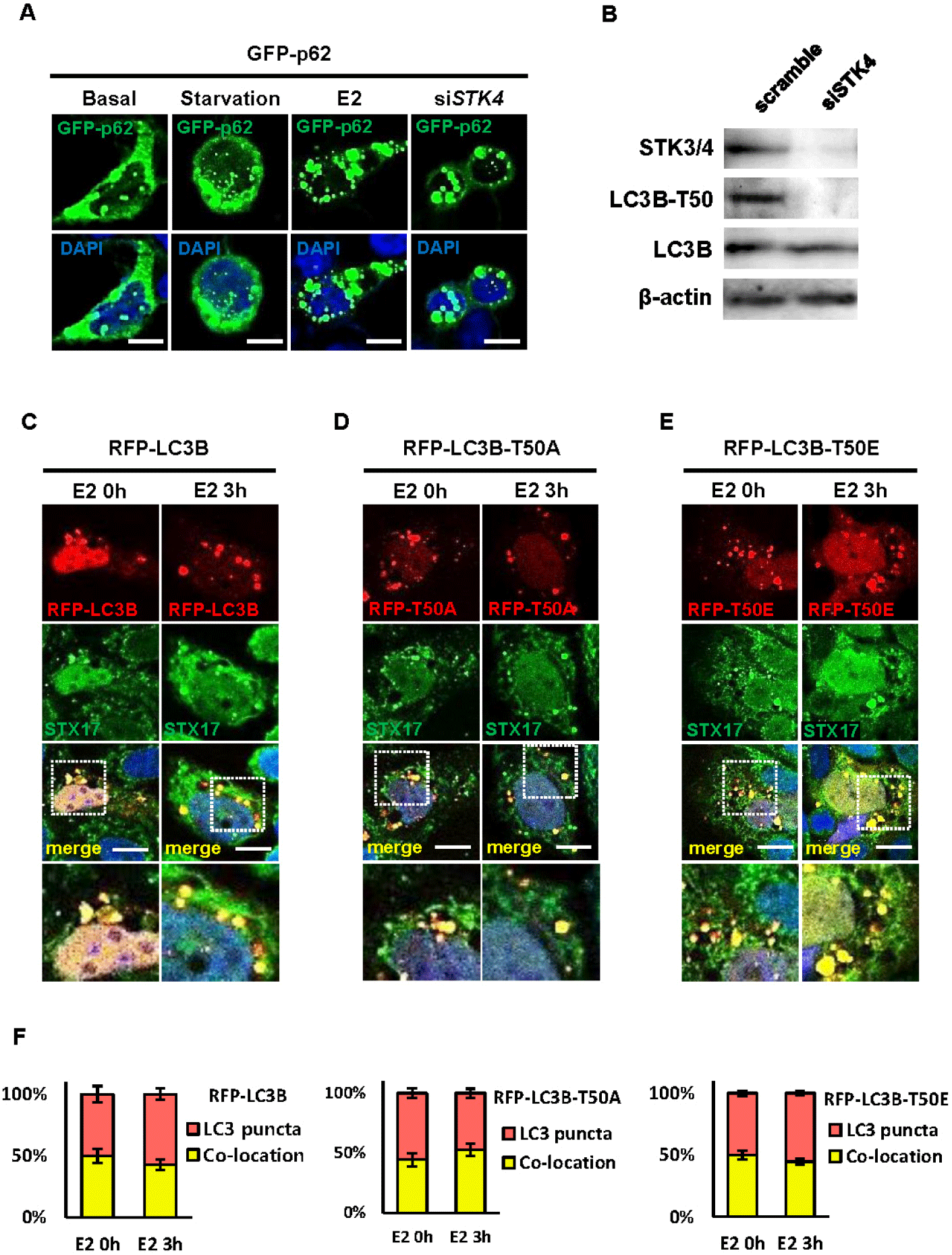
To determine the functional significance of LC3B-T50 phosphorylation, we generated two-point mutations: T50A (phospho-null mutant, replacing threonine with alanine) and T50E (phosphomimetic mutant, replacing threonine with glutamic acid). Following transient transfection of wild-type RFP-LC3B, RFP-LC3B-T50A, or RFP-LC3B-T50E into Ishikawa cells, we assessed autophagosome formation by co-staining with STX17 under E2 treatment (Fig. 4C–E). These results suggest that neither mutation affects the initial stage of autophagy (autophagosome formation) (Fig. 4F). This indicates that STK4-mediated phosphorylation of LC3B does not influence this stage, even though STK4 is directly involved in the phosphorylation process.
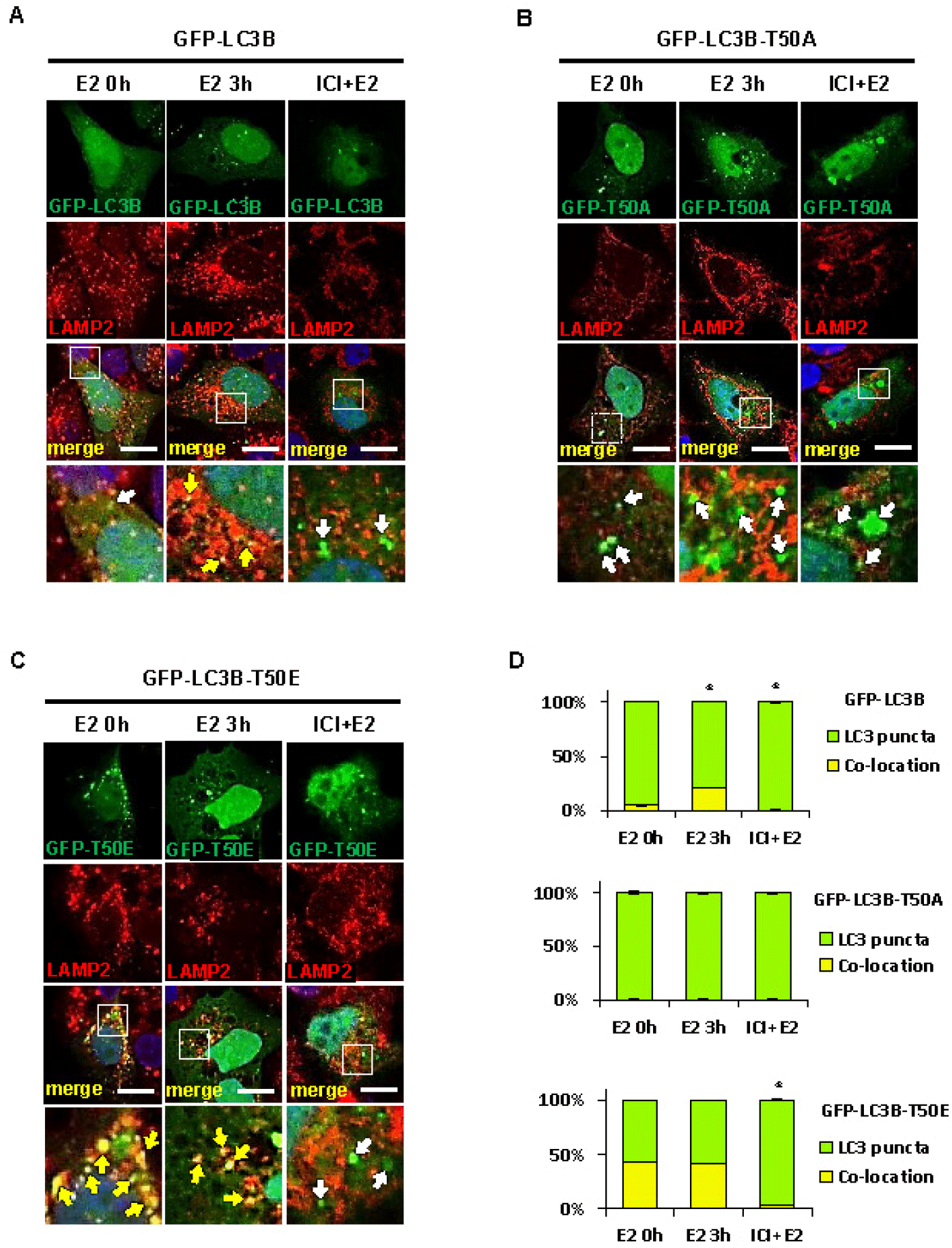
Next, we investigated whether LC3B phosphorylation affects the later stage of autophagy— autolysosome formation through autophagosome-lysosome fusion. GFP-LC3B (wild-type), GFP-LC3B-T50A (phospho-null), or GFP-LC3B-T50E (phospho-mimetic) constructs were transfected into Ishikawa cells, followed by treatment with E2 in the presence or absence of ICI 182,780. Cells were co-stained with LAMP2 to visualize autolysosomes (Fig. 5). Wild-type LC3B showed increased colocalization with LAMP2 during E2 treatment, which was blocked by ICI (Fig. 5A and D), confirming that E2 promotes autolysosome formation through estrogen receptor signaling. Strikingly, the phospho-null mutant (LC3B-T50A) failed to form autolysosomes even with E2 treatment (Fig. 5B and D), demonstrating that LC3B phosphorylation is essential for this process. Conversely, the phospho-mimetic mutant (LC3B-T50E) exhibited increased autolysosome formation even in the absence of E2, which was further enhanced by E2 treatment (Fig. 5C and D). Quantification of LAMP2-LC3B colocalization validated these immunofluorescence findings (Fig. 5D). These results demonstrate that E2 induces autolysosome formation through STK4-mediated LC3B phosphorylation. This reveals a novel regulatory mechanism that links estrogen receptor signaling with Hippo pathway kinases to regulate late-stage autophagy in the uterine epithelium.
DISCUSSION
In the present study, we investigated the regulatory role of STK4 in LC3B phosphorylation within the endometrial epithelium. We first discovered that estrogen regulates late-stage autophagic maturation in the uterine epithelium of OVX mice, as evidenced by the coordinated modulation of LC3-II levels and upregulation of fusion machinery components (Fig. 1). This regulation involves the E2-dependent expression of key fusion machinery components, including LAMP2 and STX17 (Fig. 2). We subsequently demonstrated that STK4 kinase plays an important role in the LC3B-T50. In addition, we showed that LC3B-T50 phosphorylation is a critical determinant for autolysosome maturation rather than autophagosome assembly (Figs. 4 and 5). This suggests that Hippo signaling via STK4 under estrogen is a novel mechanism for regulating autophagic maturation in the endometrial epithelium.
The endometrium changes periodically during the estrous cycle. This dynamic change in the uterus may be associated with hormonal regulation including estrogen and progesterone, as well as various unknown signaling pathways. In previous studies, we revealed that the expression of various signal factors such as RASD1 (Kim et al., 2017), AIMP1 (Jeong et al., 2015), and STK3/4 (Moon et al., 2019) may be associated with dynamic changes in the uterine epithelium through either estrogen or progesterone. Recently, we reported that STK3/4, a key component of hippo signaling pathway, is regulated in the uterus via estrogen during the estrous cycle. In the present study, we specifically targeted STK4 (MST2) via siRNA knockdown. While STK3 and STK4 are functionally redundant Hippo kinases that phosphorylate LC3B at the identical Thr50 residue (Wilkinson et al., 2015), our experimental manipulations were specific to STK4. The established functional redundancy suggests this regulatory mechanism may extend to the STK3/4 family, pending direct STK3 manipulation. In the present study, we specifically demonstrate that STK4 is required for LC3B Thr50 phosphorylation in endometrial epithelial cells. This discovery extended a new signaling component to the uterine signaling network. Interestingly, a recent report demonstrated that STK3/4 directly phosphorylates the amino acid threonine 50 of LC3, an autophagy marker in other system (Wilkinson & Hansen, 2015; Wilkinson et al., 2015). In the present study, we sought to elucidate the correlation and possibility among autophagy, estrogen, and hippo signaling in uterine endometrium.
Autophagy is an evolutionarily conserved process that is induced during cellular stress caused by a variety of conditions (Yu et al., 2018). This process prevents cell damage or death due to a lack of energy or nutrients and is a defense against various cytotoxins (Galluzzi et al., 2017). The response allows cells to adapt to an ever-changing environment. The process involves sequestering cellular material in vesicles known as autophagosomes, and delivering them to lysosomes for the digestion and recycling of ingested debris (du Toit et al., 2018). One commonly used approach for assessing autophagic maturation involves evaluating LC3 dynamics together with markers of autophagosome–lysosome fusion. Although the reduction in LC3B-II observed following E2 treatment is consistent with regulation at the late-stage autophagosome–lysosome fusion step, we cannot formally exclude the possibility that E2 may also influence LC3 lipidation or autophagosome biogenesis. Autophagic degradation activity is commonly inferred from coordinated changes in LC3 dynamics and lysosomal engagement. It is measured by determining the number of microtubule-associated protein 1 LC3 puncta. LC3 is involved in autophagosome formation and autophagy selection in the cytoplasm, and is one of the most widely used markers of autophagy (Shpilka et al., 2011; Wild et al., 2014). It is a unique component of the autophagy process. LC3 is incorporated into the newly formed autophagosome membrane and, after fusion with lysosomes, is degraded during autophagy (Nguyen et al., 2016). It is a member of the ATG8 protein family, which contains several homologues, and consists of the following isoforms: A, B, C, GABARAP, GABARAPL1, and GATE-16. Of these homologues, LC3B is a well-known factor in terms of autophagy process (Shpilka et al., 2011; Wild et al., 2014; Yu et al., 2018). Autophagy is known to occur in the cytoplasm, but LC3 is also observed in the nucleus. In fact, it is reportedly more prevalent in the nucleus than in the cytoplasm in the cell lines generally studied (Drake et al., 2010). LC3 has been shown to remain in the nucleus with respect to the high-molecular-weight complexes that constantly traverse the nucleus (Huang & Liu, 2015). Instead, nuclear LC3-positive puncta do not have autophagosomes (Buckingham et al., 2014).
Endometrial cells undergo dynamic autophagy during periodic changes to the uterus, such as the menstrual cycle. Autophagy levels have been shown to fluctuate during physiological and pathological processes in the endometrium, including menstrual shedding, tissue atrophy, and the progression of endometrial diseases (Yang et al., 2019). Conditional knockout of the autophagy protein FIP200 in the reproductive organs of female mice has been reported to reduce fertility owing to graft defects (Oestreich et al., 2020). A recent study reported that the CREBRF-mTOR-autophagy pathway plays an important role in the regulation of endometrial function in goats (Yang et al., 2018). However, there are still many unknowns regarding regulatory mechanisms of autophagy in uterine endometrium, and there is an incomplete understanding of its role in endometrial epithelial cells.
In summary, we demonstrated that late-stage autophagic maturation is regulated by estrogen and Hippo signaling in mouse endometrium. Importantly, LC3B phosphorylation by STK4 is specifically required for autophagosome-lysosome fusion but not for autophagosome formation, revealing stage-specific regulation of autophagy.
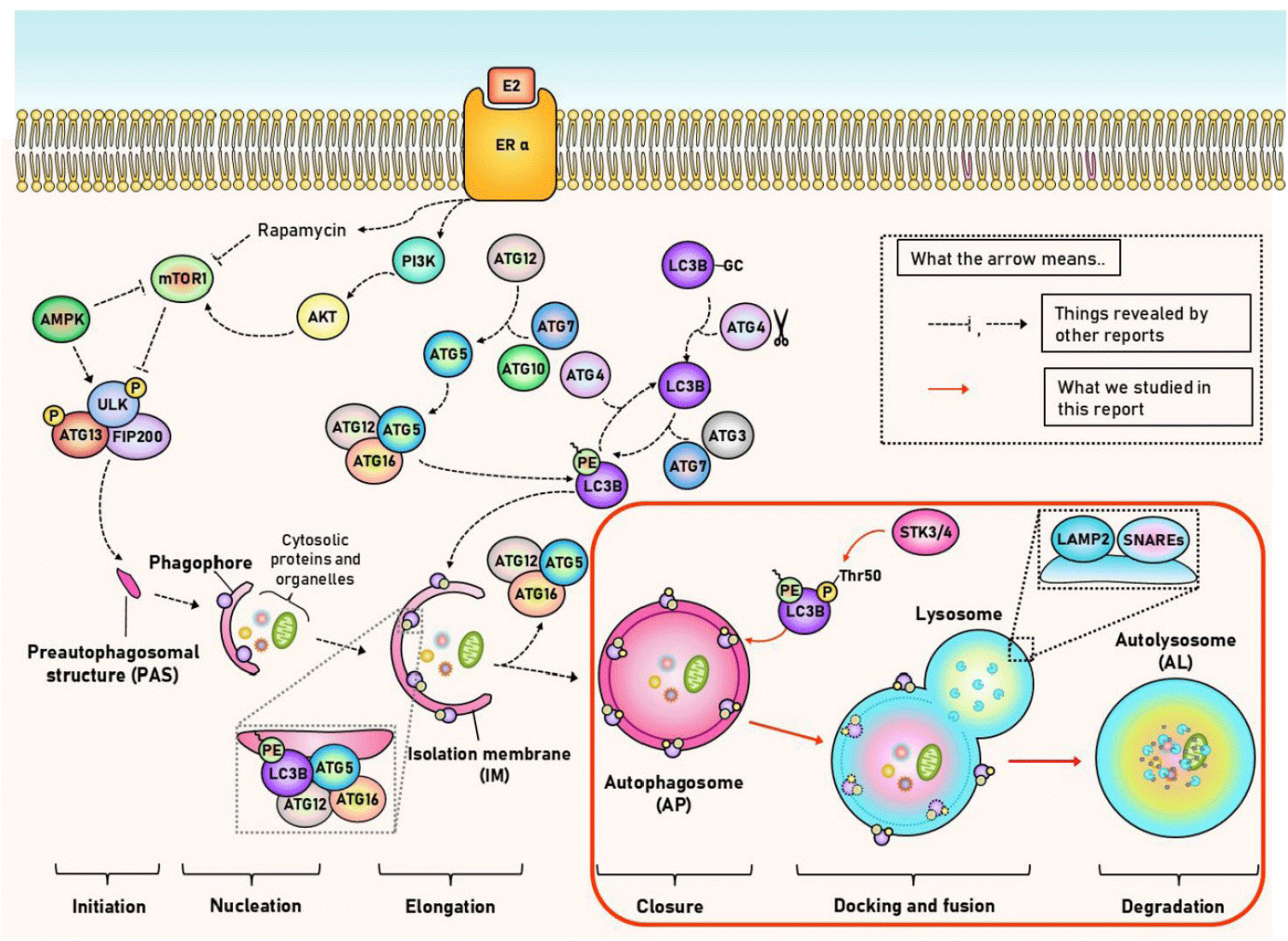
Based on our findings, we propose a working model (Fig. 6) in which estrogen signaling promotes STK4-mediated phosphorylation of LC3B-T50. This phosphorylation specifically regulates autophagosome-lysosome fusion (red boxed region) without affecting early-stage autophagosome formation. Red arrows highlight the novel estrogen-STK4-LC3B axis identified in this study, while dashed arrows represent canonical mTOR-dependent autophagy pathways.
While our data collectively support E2-regulated late-stage autophagy via STK4-mediated LC3B phosphorylation, we acknowledge that without lysosomal inhibitor (bafilomycin A1) experiments, we cannot formally exclude reduced autophagosome formation as an alternative explanation for decreased LC3B-II. Additionally, while GFP-LC3/LAMP2 colocalization indicates fusion events, tandem mRFP-GFP-LC3 reporters would provide more definitive ultrastructural resolution. These constraints define important directions for future investigation.
In conclusion, this study demonstrates that STK4 is a stage-specific regulator of autophagy in endometrial epithelium through direct LC3B phosphorylation. By revealing stage-specific regulation of autolysosome formation without affecting autophagosome formation, we identify a novel mechanism distinguishing hormone-regulated from nutrient-starvation-induced autophagy. These findings establish important crosstalk between estrogen receptor and Hippo signaling pathways, providing new insights into the complex signaling networks governing uterine physiology.