
INTRODUCTION
Colorectal cancer (CRC) is the third most common type of cancers and over millions of patients has been diagnosed annually. Main curative treatment for CRC primarily depends on surgical resection followed by radiation and chemotherapy. This treatment, however, often results in unsatisfactory outcome especially for the patients undergoing metastasis and in advanced stages, which requires the development of new therapeutic approaches (Wolpian & Mayer, 2008).
Mutations in CRC are frequently found in protooncogenes encoding RAS (approximately 40%), PI3K (15–30%) and BRAF (15%), which in normal cells stimulate the cellular growth and proliferation in response to growth factors (Laurent-Puig et al., 2009). The activating mutations of these oncogenes results in aberrant cell proliferation via a series of signaling casades, mainly the RAS/RAF/MEK/ ERK and PI3K/Akt/mTOR pathways, which thus play important roles in the regulation of malignant cell proliferation (Mendoza et al., 2011). Therefore, development of inhibitors targeting these pathways has become an attractive therapeutic strategy and such molecular targeting drugs have been actively searched and tested clinically and preclinically. One of such agents is the imidazoquinoline derivative BEZ235, a novel and orally bioavailable dual inhibitor of PI3K and mTOR. This compound inhibits PI3K and mTOR kinase activities by binding to the ATP binding cleft of these enzymes (Maira et al., 2008) and is currently being evaluated in phase I/II clinical trials in patient with solid tumors including CRC (Roper et al., 2011; Blaser et al., 2012).
Most cancer therapeutic approaches, either chemotherapy or targeted therapy, have focused on induction of cell death and inhibition of cell growth. Resistance to anticancer drugs, however, compromises treatment outcome and limits the effectiveness of cancer treatment (Szakacs et al., 2006). Drug resistance can result from a variety of mechanisms such as tumor cell heterogeneity, drug efflux, metabolism, autophagy induction, and genetic and epigenetic alterations as a cellular response to drug exposure (Sui et al., 2013). Autophagy is a lysosomal catabolic process which is essential for regulation of cell survival and death to keep cellular homeostasis. Substantial evidence suggests that autophagy has dual roles in cancers, acting as either prosurvival or prodeath mechanism against chemotherapy treatment, which contributes to the anticancer efficay as well as drug resistance (Shigemitsu et al., 1999; Takeuchi et al., 2005; Sui et al., 2013). Many anti-cancer drugs including targeted therapies induce autophagy as a protective and prosurvival mechanism and blockage of autophagy thus augments the cytotoxic effect of chemotherapeutic agents (Li et al., 2013b). On the other hand, excessive and persistent autophagy may induce autophagic cell death following chemotherapeutic agents (Li et al., 2013a). One of the main regulators of autophagy is mTOR, and inhibitors of mTOR such as rapamycin and BEZ235 are reported to induce autophagy in urothelial and renal cancer cell (Li et al., 2013a, 2013b). Although previous studies have showed that inhibition of autophagy potentiates the anticancer effect of targeted therapy in CRC (Yang et al., 2010), there is no report to examine the effectiveness of a novel dual PI3K/mTOR inhibitor BEZ235 as an anticancer drug in relation to autophagy induction in CRC cell.
The present study, therefore, was designed to elucidate the effects of BEZ235 on cell viability, cell cycle distribution, apoptosis and autophagy induction using human CRC cell line HCT15. In addition, we examined whether inhibition of autophagy by autophagy inhibitor chloroquine (CQ) may potentiate or decrease the anticancer activity of BEZ235 in HCT15 CRC cells.
MATERIALS AND METHODS
The human colorectal cancer cell line HCT15 was obtained from the American Type Culture Collection (Manassas, VA). Cells were grown in a monolayer culture in DMEM (Sigma) supplemented with 10% fetal bovine serum (Sigma) and 1% streptomycin/penicillin, and were maintained at 37°C in a humidified atmosphere consisting of 5% CO2 and 95% air. Cells were regularly tested for mycoplasma contamination by treating 5 μg/mL of Plasmocin (InvivoGen). BEZ235 was purchased from LC laboratories (Woburn, MA). CQ, acridine orange, 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) were all purchased from Sigma.
To determine the effects of BEZ235 on HCT15 cell viability, we used MTT assay as described previously (Park & Lee., 2014). Cells were harvested and seeded at 5 × 104 cells per well (0.5 mL) in 24-well plates and incubated overnight at 37°C. Cells were then incubated for 1d, 2d and 3d with BEZ235 to determine the concentration- and time-dependent cell survival rate, or for 48 hours with CQ and/or BEZ235 to assess the effect of autophagy inhibition on BEZ235-induced cell viability. Then, cells were incubated with MTT reagent for 3 hours at 37°C, followed by solubilization of the formazan crystal with propanol for 30 minutes. Absorbance was measured at 570 nm with a microplate analyzer. All the experiments were performed in triplicate.
Western blot assays were performed as described previously (Song et al., 2011) using the following antibodies: cyclin D1, cyclin A, cyclin B1, ERK, pERK (Tyr204), pAkt (Ser473), Akt (Santa Cruz Biotechnology, Santa Cruz, CA), LC3, cleaved caspase-3 (Cell Signaling, Beverly, MA). β-actin (Cell Signaling) was used as loading control following by goat-antimouse IgG or goat-antirabbit IgG conjugated to horseradish peroxidase (Cell Signaling). Enhanced chemiluminescence was used for detection (Santa Cruz Biotechnology).
HCT115 cells were plated onto 60 mm dishes and treated with various concentrations of BEZ235 (0, 20, 50, 100, 500 1,000 nM) for 24 hr. Cells were harvested by treating trypsin-EDTA for 5 min at 37°C, washed with PBS and fixed overnight in 50% ethanol at 4°C. Fixed cells were washed and incubated with RNase (200 μg/mL) for 30 min at 37°C, and followed by propidium iodide (PI) staining. Cells were analyzed using BD FACSCalibur Flow Cytometry System and data were analyzed using CellQuest software.
The characteristics of autophagy is the formation of acidic vesicular organelle (AVO) which consist predominantly of autophagosome and autophagolysosome. To visualize the development of AVOs after BEZ235 treatment, we performed vital staining with acridine orange. Briefly cells were washed twice with DPBS, stained with acridine orange (1 μg/mL) in HBSS containing 5% FBS for 15 minutes and then observed with a fluorescence microscope.
RESULTS
To delineate the antiproliferative activity of BEZ235 in HCT15 CRC cells, cells were incubated with various concentrations of BEZ235 (2.5, 5, 10, 20 and 100 nM) for 1d, 2d and 3d, and cell viability was determined using MTT assay. As shown in Fig. 1A, BEZ235 inhibited the cell viability in concentration- and time-dependent manner. The IC50 levels at the time points of 1d, 2d and 3d incubation were 235.8 nmol/L, 75.9 nmol/L, and 31.5 nmol/L, respectively. Next, we investigated whether the observed decrease in cell viability after BEZ235 treatment is linked to the perturbation of signaling pathways for cell survival and proliferation. Western blot analysis to monitor the alteration of Akt and ERK activity, main components of PI3K/Akt/mTOR and RAS/RAF/MEK/ERK signaling pathways, was performed for the cells treated with BEZ235 at 20 to 1,000 nmol/L for 24 hrs. As revealed in Fig.1B, the phosphorylation of Akt at Ser473 was increased in concentration-dependent manner and phosphoryation of ERK at Tyr204 was remarkably suppressed in all the time points of treatment.
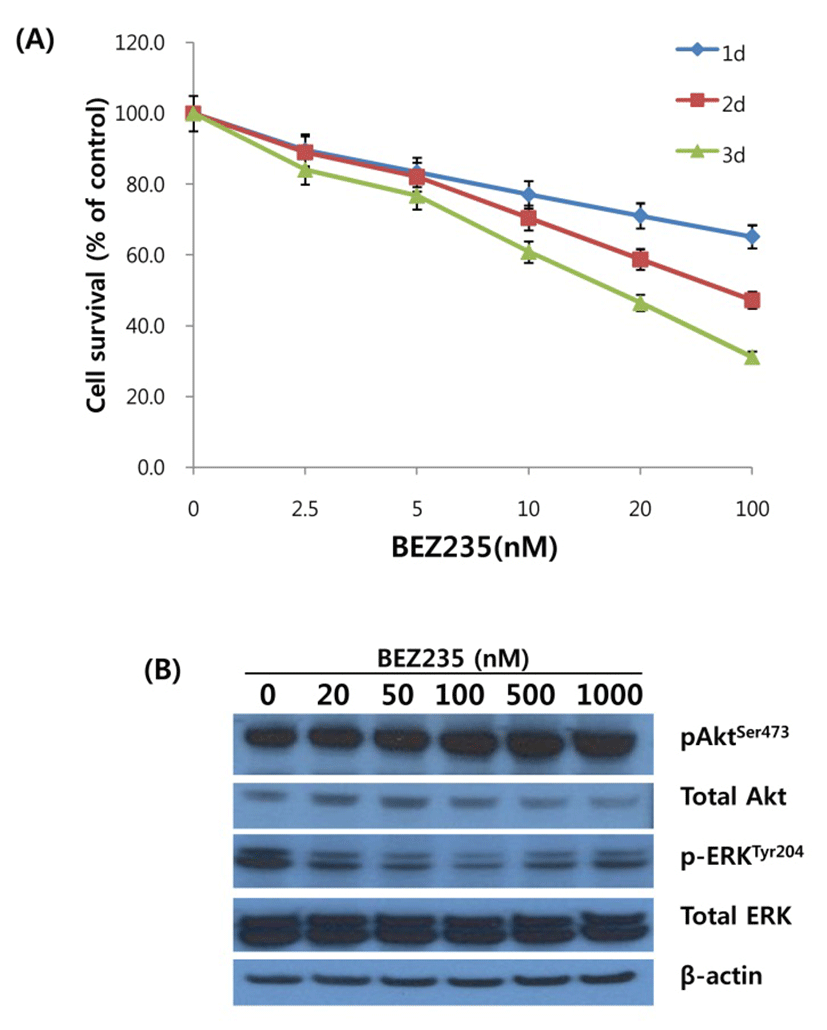
On the basis of antiproliferation effect of BEZ235 on HCT15 cells, we next screened the cell cycle distribution in response to BEZ235 treatment. Cell cycle analysis of HCT15 cells by flow cytometry using PI staining is shown in Fig. 2A. Consistent with the inhibition of cell viability following BEZ235 treatment, the percentage of HCT15 cells in G1 phase was increased in a concentration-dependent manner from 56.9% of control to 83.5% of 1,000 nmol/L. The increase of cell population in G1 phase is accompanied by a reduction of that in S and G2/M phases, indicating that BEZ235 induces G1 arrest of cell cycle. In contrast, the marginally increased level of sub-G1 population characteristic of apoptosis was observed following 500 nM and 1,000 nM BEZ235 treatment. To further confirm that the antiproliferative activity of BEZ235 is mediated by G1 cell cycle arrest, the expression of proteins which participate in the regulation of cell cycle were screened by Western blot. As found in Fig. 2B, whereas there is no obvious alteration in cyclin D1 after 24 hr treatment of BEZ2235, it caused a remarkable reduction in the expression levels of cyclin A and cyclin B1. Western blot for cleaved caspase-3 showed the slight increased levels of expression with 500 and 1,000 nM BEZ235 treatment, which is consistent with the result of cell cycle analysis.
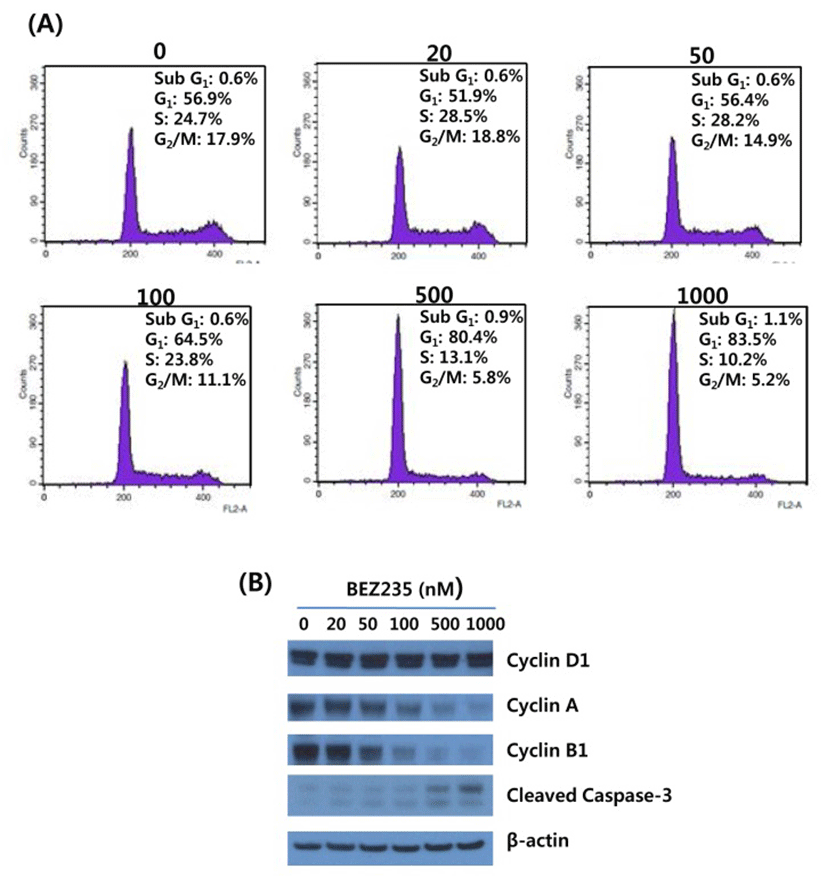
Substantial evidence has shown that mTOR is a negative regulator of autophagy, and most of chemotherapeutic and targeted agents used for cancer therapy induce autophagy in cancer cells (Shigemitsu et al., 1999; Takeuchi et al., 2005). Thus, we investigated whether BEZ235 treatment induces autophagy in HCT15 CRC cells by using acridine orange (AO) staining and LC3-I conversion to LC3-II as autophagy markers. The lysosomotropic agent AO accumulates in acidic vesicular organelles (AVOs), a marker of onset of autophagy. When excited with blue light, AO emits red fluorescence in AVOs and green fluorescence in cytoplasm and nucleus (Takeuchi et al., 2005). During autophagy the cytosolic form of LC3-I (16 kDa) is converted to the lower migrating autophagosome-bound form of LC3-II (14 kDa) through lipidation to be associated with autophagic vacuoles (Li et al., 2013a).
As revealed in Fig. 3, BEZ235-treated cells showed strong and abundant cytoplasmic red fluorescence that reflects the development of AVOs and autophagy induction, whereas untreated cells exhibited little cytoplasmic staining. Analysis of LC3 lipidation by Western blot revealed that BEZ235 caused a marked increase of lipidated form LC3-II accompanied by a decrease of the non-lipidated LC3-I (Fig. 3B). Together, these data indicate that BEZ2325 induces autophagy in human HCT15 CRC cells.
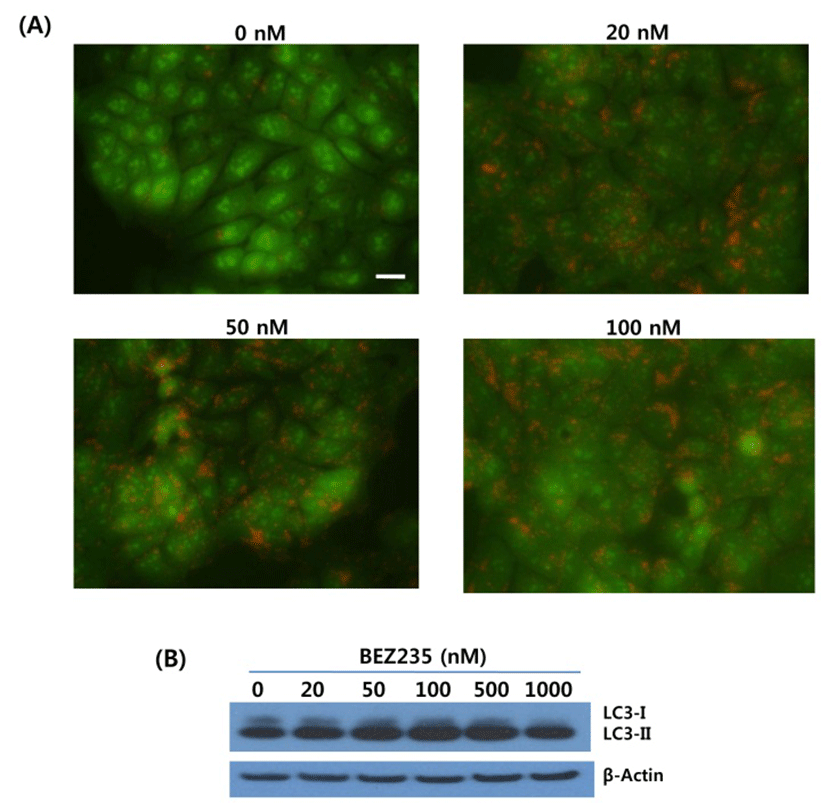
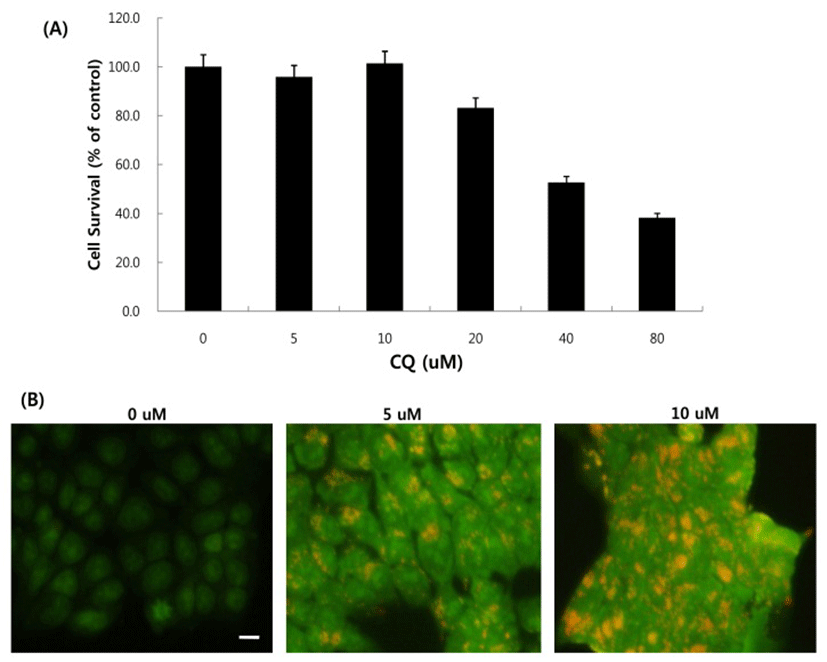
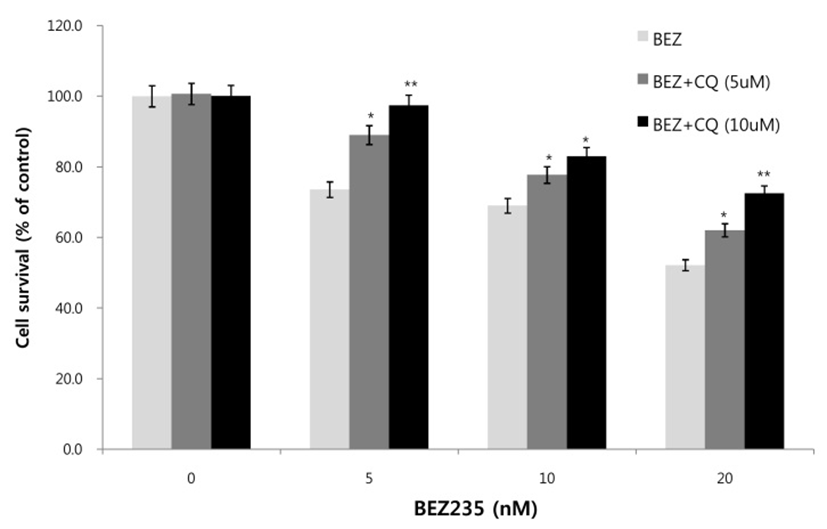
Next, we asked whether inhibition of autophagy affects the growth inhibition in response to BEZ235 treatment. To this end, we used a lysosomotropic agent chloroquine (CQ), which inhibits the fusion of autophagosomes with lysosome and thus commonly employed as an autophagy inhibitor (Mizushima et al., 2010). Recently, however, several reports suggest that high doses of CQ can promote lysosomal membrane permeabilization, resulting in lysosome-mediated apoptosis (Seitz et al., 2013; Park & Lee, 2014). Therefore, we determined the dose of CQ which effectively inhibits autophagy without affecting the cell viability of HCT15 cells. As shown in Fig. 4A, CQ at 5 and 10 uM did not affect cell viability while CQ above 20 uM resulted in decreased cell survival compared with control. CQ has been known to induce the accumulation of AVOs since CQ inhibits the late step of autophagy blocking the fusion of autophagosome and lysosome (Sasaki et al., 2010). In the present study, AO staining following CQ (5 and 10 μM) treatment showed the abundant red fluorescent AVOs, further confirming that CQ inhibits autophagy (Fig. 4B). In the experiment to assess the effect of autophagy inhibition on cell viability in response to BEZ235 treatment, cells were co-treated with BEZ235 plus CQ. Fig. 5 shows that the combination of BEZ235 and CQ resulted in rescued cell viability, and the rescued effect was more pronounced in HCT15 CRC cells following treatment with 10 μM CQ than 5 μM CQ. Thus, it is plausible that autophagy induced by BEZ235 treatment acts as an inhibitory mechanism of cell proliferation and blockage of autophagy rescues the cell viability inhibited by BEZ235 treatment in HCT15 CRC cells.
DISCUSSION
Molecular targeting for signaling pathways offers new therapeutic options in the treatment of CRC. During the past decade, several new agents have been developed and screened in a number of cancer types in both preclinical models and clinical trials. In the present study using HCT15 CRC cells, we first examined the antiproliferative activity of BEZ235 and the effects of its treatment on the activity of AKT and ERK, the key components of two signaling pathways, PI3K/Akt/mTOR and RAS/RAF/MEK/ERK, involved in cell survival, proliferation, apoptosis and differentiation. We found that BEZ235 effectively inhibited the growth of this CRC cell line with IC50 of nanomolecular concentrations as demonstrated in other types of cancer cells (Maira et al., 2008; Xu et al., 2011). On the contrary to reports using other tumor cells (Xu et al., 2011; Li et al., 2013b), BEZ235 caused the marked increase of phosphoryalated Akt at Ser473 and suppressed the phosphoryation of ERK at Tyr204. mTOR protein exists in two distinct complexes, mTORC1 and mTORC2 which are both inhibited by BEZ235. mTORC1 and mTORC2 phosphorylate Akt at Thr308 and Ser473, respectively. Frequently, however, inhibition of mTORC1 is linked with an activation of mTORC2 complex, which leads to subsequent phosphorylation of Akt at Ser473 (Schult et al., 2012; Kuger et al., 2015). Several studies also suggested that mTORC2 promotes the phosphorylation of Akt at Ser473 and this feedback loop serves as a rescue mechanism of cells in response to mTOR kinase inhibition (Sarbassov et al., 2005; Guertin et al., 2006). The decrease of ERK phosphorylation following BEZ235 treatment in the present study can result from the negative regulation between two signaling pathways. Such cross-inhibition is often found when one pathway is chemically blocked, hence releasing cross-inhibition and efffectively activating the other pathway (Mendoza et al., 2011).
Numerous studies demonstrated the association of cell cycle arrest with cancer therapeutic applications, and the inhibition of cell cycle has become a potential target for the treatment of cancer (Lee & Sicinski, 2006; Shapiro, 2006). Progression of cell cycle is regulated by reversible phosphorylation induced by cyclin-dependent kinase (CDK) and its essential activating partner, cyclin. Cyclin D is the first cyclin to rise when cells approaches the G1 checkpoint. Association of cyclin D with its catalytic partners, CDK4 and CDK6, induces the synthesis of cyclin E, followed by cyclin A synthesis in late G1 phase and S phase. Cyclin B accumulates during G2 phase and prepares the cells for entry into mitosis and executes the early steps of mitosis. Our present study shows that the antiproliferative activity of BEZ235 results mainly from G1 phase arrest of cell cycle with marginal effect on apoptosis in HCT15 CRC cells. In addition, the expression level of cyclin D was not altered after BEZ235 treatment, whereas the marked decrease of cyclin A and B1 levels was observed in dose-dependent manner. Several studies with other cancer cell lines suggested that growth inhibitory effect of BEZ235 is attributed to G1 arrest with the reduction of cyclin D expression (Yang et al., 2013; Moon et al., 2014), and cyclin A has been regarded as a S phase cyclin and its suppression induces S phase arrest in the cell cycle (Joe et al., 2002; Hung et al., 2013). The temporal relationship between cyclin A accumulation and DNA replication, however, showed that cyclin A accumulates precisely at the G1/S border and DNA replication does not take place before its accumulation in cancer cells (Erlandsson et al., 2000). Therefore, considering that flow cytometry for the cell cycle analysis is based on DNA content within cell, the suppression of cyclin A seems to result in the increase of cell population in G1 phase not in S phase. Unaltered cyclin D expression seen in this study appears to result from the increased phosphorylation of Akt in response to inhibition of negative regulation by mTOR following BEZ235 treatment, which is in agreement with other report that Akt activation is associated with expression of cyclin D (Muise-Helmericks et al., 1998). Together, our results suggest that antiproliferative effect of BEZ235 on HCT15 CRC cells appears to be due to G1 arrest of cell cycle via inhibition of cyclin A expression.
Recently, several groups already reported that BEZ235 induces autophagy in diverse cancer cell lines (Xu et al., 2011; Li et al., 2013a; Li et al., 2013b). In the present study, we also found that BEZ235 causes significant autophagy in HCT15 CRC cells, evidenced by detection of increased levels of LC3B-II, a cleaved product of LC3B-1, and numerous AVO development by vital staining with AO. Considering that mTOR acts as a negative regulator of autophagy, it is not surprising to find induction of autophagy in cells treated with BEZ235, a dual PI3K/ mTOR inhibitor. Although it was suggested that many anticancer drugs used in chemotherapy and molecular targeted therapy stimulate autophagy, the role of autophagy on cell survival or cell death is controversial Sui et al., 2013). In our study, autophagy induced by BEZ235 appears to be cell destructive, since the combination treatment of BEZ235 with the autophagy inhibitor CQ counteracts the antiproliferative effect seen in treatment of BEZ235 alone (Fig. 5). At present, the mechanism of this rescued cell viability upon autophagy inhibition is unknown. Apart from its apparent survival role, autophagy has also been suggested as having a cell destructive role by mediating autophagic cell death or type II physiological cell death, which is regulated by a molecular mechanism different from that of apoptosis (Li et al., 2013a). Recently, cancer cells harboring activating mutations in RAS have been reported to have high basal levels of autophagy, which is required to maintain active tumor growth (Guo et al., 2011). Furthermore, the expression of oncogenically mutated ras gene triggers cellular degeneration with biochemical and morphological features typical of autophagic cell death (Chi et al., 1999). The excessive and persistent induction of autophagy has shown to lead to autophagic cell death under certain circumstances (Li et al., 2013b). Thus, we tentatively suggest that high basal level of autophagy in HCT15 cells bearing RAS mutation and additional induction of autophagy in response to BEZ235 treatment result in autophagic cell death, and inhibition of autophagy with CQ treatment counteracts the the antiproliferative effect of BEZ235 on HCT15 CRC cells.
In summary, the present study using HCT15 CRC cells showed that BEZ235 effectively inhibits cell proliferation by G1 arrest of cell cycle, which occurs via suppression of cyclin A expression. In addition, BEZ235 induces autophagy and inhibition of autophagy leads to the rescued cell viability, indicating that autophagy acts as cell destructive. Our findings provide novel insights into the role of autophagy as a cell death mechanism in the area of CRC therapeutics.