
INTRODUCTION
Leydig cells are located in the interstitial compartment of the testis and produce testosterone (T), which is required to maintain male physiology and for reproduction (i.e., spermatogenesis) (Roberts & Zirkin, 1991). Leydig cells in adult testes generally do not proliferate under normal physiological conditions. Hence, they fully differentiate into different cell types (Ewing & Zirkin, 1983; Zirkin & Papadopoulos, 2018). Furthermore, Leydig cells are known to undergo aging and/or senescence throughout life (Chen & Hardy, 1994; Chen et al., 2009). To achieve T production, a series of cellular organelles participate in steroidogenesis in Leydig cells, which include lipid droplets, mitochondria, and the smooth endoplasmic reticulum (sER). During steroidogenesis, cholesterol supply, pregnenolone synthesis, and cell-specific steroid production subsequently occur in these cellular organelles (Miller, 1995; Miller & Bose, 2011). Among these, most steroidogenic proteins and enzymes are localized and enriched in the mitochondria and sER (Stocco & Clack, 1996; Miller, 2008; Miller, 2011). Thus, these cellular organelles have suitable conditions for adequate T production in Leydig cells.
Autophagy involves either degradation and/or clearance of dysfunctional/damaged cellular organelles or the reuse of intracellular elements (Klionsky & Emr, 2000; Mizushima & Komatsu, 2011). In general, autophagosomes consisting of double membrane-bound structures mature and fuse with lysosomes to achieve their function. Autophagy is classified into three forms: macroautophagy, microautophagy, and chaperone-mediated autophagy (Schnebert et al., 2022). Macroautophagy is performed via the autophagosome-lysosome fusion complex. The representative ultrastructural features of autophagic compartments include the phagophore, autophagosome, and autolysosome. Autophagy can be detected via immunocytochemical staining of the microtubule-associated protein light chain 3 (LC3)-II or observed via emission of green fluorescent protein (GFP)-tagged LC3 (Klionsky et al., 2008; Levine & Kroemer, 2008). Experimentally, autophagy can be induced via inhibiting the mammalian target of rapamycin kinase with rapamycin (Noda & Ohsumi, 1998) or blocked via inhibiting the class III PI3K with 3-methyladenine (3-MA) (Seglen & Gordon, 1982). Although autophagy was originally recognized as a survival response to starvation and other forms of cellular stress, it is now considered an important cellular phenomenon directly associated with cell death, cell survival, differentiation, cellular homeostasis, and cell senescence and aging (Tsujimoto & Shimizu, 2005; Glick et al., 2010; Mizushima & Komatsu, 2011). Many human diseases, including neurodegeneration, cancer, and diabetes are associated with defects in autophagy (Choi et al., 2013; Levine & Kroemer, 2019).
Therefore, in this study, we investigated the involvement of autophagy with steroidogenesis in testicular Leydig cells. To monitor autophagy, LC3 protein alteration was examined in Leydig cells after exposure to gonadotropin and the extent of autophagy was comparatively analyzed with the expressions of steroidogenic proteins and T production. Changes in steroidogenic activity in Leydig cells were also inspected in the absence or presence of autophagy inhibitors.
MATERIALS AND METHODS
Dibutyryl cyclic-AMP (dbcAMP), human chorionic gonadotropin (hCG), dimethyl sulfoxide, ploy-L-lysine solution, Dulbecco’s Modified Eagle’s Medium/Nutrient Mixture (DMEM) F-12 Ham, medium 199, Percoll, gentamicin and puromycin were purchased from Sigma Chemical (St. Louis, MO, USA). Complete protease inhibitor cocktail tablet was from Roche Applied Science (Mannheim, Germany). Horse serum was from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). MitoTracker™ Red CMXRo fluorescent dyes was from Molecular Probes (Thermo Fisher Scientific). Lipofectamine™ 2000 Transfection Reagent was obtained from Invitrogen (Thermo Fisher Scientific). P4 and T enzyme-linked immunosorbent assay (ELISA) kits were purchased from IBL (Hamburg, Germany). The western enhanced chemiluminescence (ECL) detection reagent was purchased from Bio-Rad (Hercules, CA, USA). Anti-LC3 antibody was purchased from Novus Biologicals (Littleton, CO, USA). Anti-StAR and actin antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-CYP11A1 antibody was from Chemicon (Millipore, Billerica, MA, USA). Rabbit and mouse IgG-conjugated with horseradish peroxidase were purchased from Cell Signaling Technology (Danvers, MA, USA).
For hCG-injection experiment, immature male Sprague-Dawley (SD) rats (3 weeks of age) were purchased from SamTako Bio-Korea (Osan, Korea). The rats were housed in a climate-controlled (21±2°C) animal room at a constant 12-h light/dark cycle with free access to feed and water ad libitum. All procedures were performed in accordance with protocols approved by the Dong-A University Animal Care and Use Committee. The rats were given an intraperitoneal injection of diluent (phosphate-buffered saline, PBS) or hCG (30 IU). After 12 h and 48 h of injection, the rats were sacrificed by carbon dioxide asphyxiation and whole blood was collected by cardiac puncture. The testes were dissected, the testes were used to collect interstitial fluid (IF) and to isolate Leydig cells. Interstitial testicular fluids (ITF) was collected according to a previously described method (Turner et al., 1984). Blood was allowed to clot at room temperature (RT) and centrifuged at 3,000μg for 10 min at 4°C. The testes of adult male SD rats (16 weeks of age) were used for isolation of primary Leydig cells and employed for their culture experiments.
Primary Leydig cells were isolated from the testes as described previously (Klinefelter et al., 1987), with slight modifications. In brief, testes were decapsulated and incubated with dissociation buffer containing collagenase (0.25 mg/mL) at 34°C for 30 min. Dissociated testes were filtered through a 100-μm pore size nylon mesh to remove the seminiferous tubule mass. The filtrate was centrifuged at 800μg for 5 min at RT and the pellet was resuspended in Percoll solution, which is mixed with Percoll buffer and iso-osmotic Percoll in Hanks′ Balanced Salt solution (HBSS). After centrifugation (20,000μg, 60 min, 4°C), the supernatant was discarded and the cell pellets were washed with dissociation buffer. The isolated Leydig cells were resuspended in M-199 medium containing 15 mM HEPES, 0.1% BSA, 5 μg/mL gentamicin, 50 U/mL penicillin, and 50 μg/mL streptomycin, which yielded 90% pure Leydig cells. For hCG treatment experiments, the purified cells (1.0×106) were plated onto a 6-well plate and cultured without serum at 34°C in a humidified incubator with an atmosphere of 5% CO2.
MA-10 cells (provided by Dr. M. Ascoli from the University of Iowa, USA) were cultured with DMEM/HAM’s nutrient mixture F-12 modified to contain 1.2 g/L of NaHCO3, 20 mM HEPES, 50 μg/mL gentamicin, and 15% heat-inactivated horse serum (pH 7.4) at 37°C in a humidified atmosphere with 5% CO2. Cells were maintained on 0.1% gelatin-coated culture dishes. The cells (5×105 cells) were seeded in gelatin-coated 6-well plate and dbcAMP was treated at confluence with approximately 50%–60%. For EGFP-LC3 puncta staining, MA-10 cells grown on coverslips and transfected with EGFP-LC3 with 0.1% lipofectamine 2000 reagent for 24 h were cultured in the absence or presence of dbcAMP.
T or P4 concentration was determined in serum, ITF or culture medium using a corresponding ELISA kit, following the manufacturer’s instructions.
The cultured or isolated Leydig cells were washed with cold PBS and resuspended in lysis buffer [300 mM NaCl, 0.5% Triton X-100, 50 mM Tris-HCl (pH 7.4), 25 mM NaF, 1 mM Na3VO4, 10mM Na4P2O7, and protease inhibitor] for 40 min on ice. The lysates were centrifuged at 13,000μg at 4°C for 20 min, the supernatants were collected and protein concentration was measured using the bicinchoninic acid protein assay kit. 20 μg of protein extract with sodium dodecyl sulfate (SDS)-loading buffer was electrophoretically separated on a 8%–15% gradient SDS-PAGE gel, and transferred onto a nitrocellulose membrane. The membranes were blocked with 5% non-fat dry milk dissolved in Tris-buffered saline buffer containing 0.05% Tween-20 at RT for 1 h. The blots were incubated with primary antibodies, followed by incubation with appropriate HRP-conjugated secondary antibodies. The signals were detected with ECL detection reagent in the LAS-4000 (Fuji, Tokyo, Japan) at the Neuroscience Translational Research Solution Center (Busan, Korea).
Data were expressed as the mean±SD of at least 3 independent experiments. The difference in means between 2 groups was analyzed using the Student’s t-test. For multiple group comparisons, one-way ANOVA followed by Tukey’s test was performed by GraphPad Prism software (version 4.0; GraphPadc, San Diego, CA, USA). Mean values were considered significantly different at p<0.05.
RESULTS AND DISCUSSION
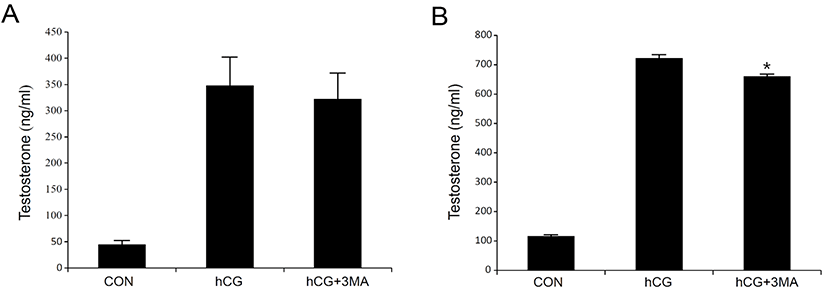
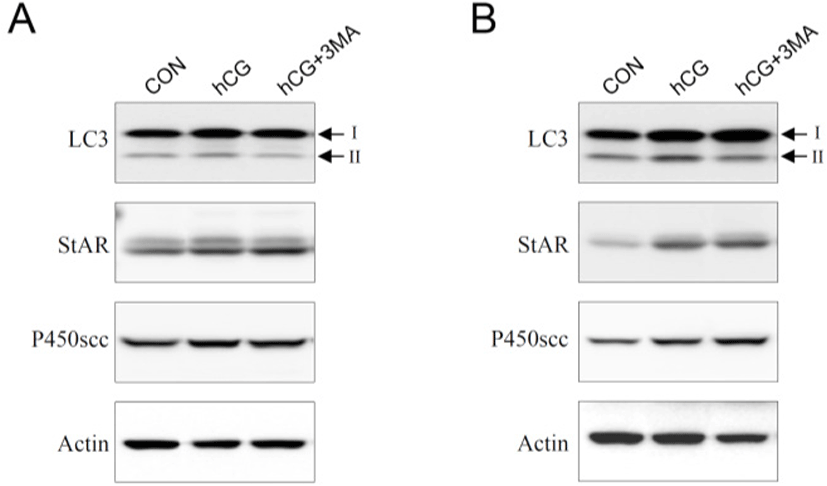
Most steroidogenic cells exhibit common ultrastructural characteristics. Compared to other cell types, they retain a higher number of lipid droplets, mitochondria, lysosomes, sER, along with a larger and circular nucleus. Except for the nucleus, the cellular organelles are degraded, cleaned, recycled, and/or renewed to maintain homeostatic integrity. Autophagy is a cellular mechanism specifically responsible for these processes (Klionsky & Emr, 2000; Mizushima & Komatsu, 2011). It is thought to be particularly important in differentiated or aging cells because of their comparatively longer life span. Steroidogenic Leydig cells are highly differentiated and undergo aging (Ewing & Zirkin, 1983; Chen & Hardy, 1994). Thus, we hypothesized that autophagy may play an important role in regulating the steroidogenic activity and/or efficacy of the Leydig cells. To test this hypothesis, primary Leydig cells isolated from adult rat testes were incubated with hCG in the absence or presence of 3-MA, an autophagy inhibitor. This resulted in an increased production of T in a time-dependent manner (Fig. 1A and B). However, hCG-stimulated T production was not phenomenally altered in the presence of 3-MA (Fig. 1A and B). Although pretreatment with 3-MA demonstrated a tendency to decrease hCG-induced T production, the differences were significant only at a higher time point of 24 h following hCG treatment (Fig. 1B). Under similar conditions, changes in cellular protein content associated with autophagy (LC3) and steroidogenesis [steroidogenic acute regulatory protein (StAR) and cytochrome P450 side chain cleavage (P450scc)] were monitored using western blot analysis (Fig. 2). In cells involved in autophagy, LC3 exists in two forms: an 18-kDa cytosolic protein (LC3-I) and a processed 16-kDa protein (LC3-II). Notably, LC3-II was present in the control (vehicle-treated) cells in all the experiments (Fig. 2A and B). On the other hand, their expression increased in comparison to the control, at both 3 and 24 h following hCG treatment, while pretreatment with 3-MA suppressed this elevation (Fig. 2A and B). However, the hCG-induced increase in StAR and P450scc protein levels were not significantly altered by 3-MA treatment (Fig. 2A and B). These results indicate that autophagy might be a basic phenomenon for maintaining Leydig cell homeostasis and function, including the modulation of steroidogenic activity.
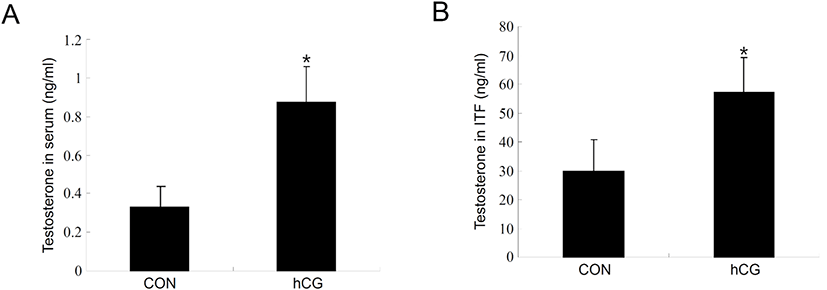
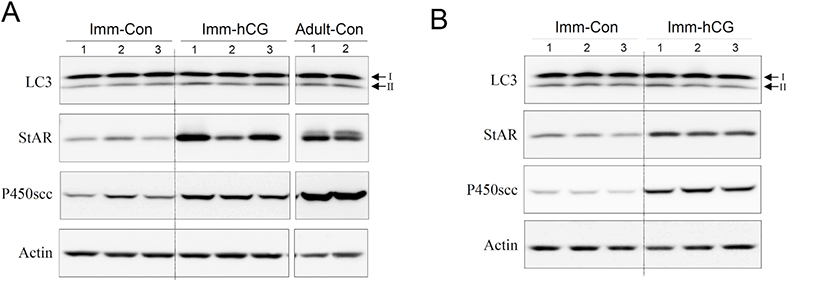
Next, we set up an hCG-administered immature rat model to monitor changes in the process of autophagy of immature Leydig cells in vivo. Administration of hCG to immature rats successfully increased T levels in both serum (Fig. 3A) and intratesticular fluids (Fig. 3B). Leydig cells isolated from immature rat testes 12 h following hCG treatment demonstrated relatively increased levels of LC3-II protein compared to the control group (Fig. 4A). Furthermore, LC3-II levels shown in these cells reached almost the identical to those from normal adult testes (Fig. 4A). However, LC3-II protein levels were almost comparable or even slightly lower than the controls at 48 h following hCG treatment (Fig. 4B). Expression of StAR and P450scc was consistently upregulated at both 12 and 48 h after hCG treatment (Fig. 4A and B). These results imply that although steroidogenesis does not occur extensively in immature Leydig cells, autophagic activity still occurs at a certain level in these cells and that relatively increased autophagic activity is accompanied by early initiation of steroidogenesis in Leydig cells.
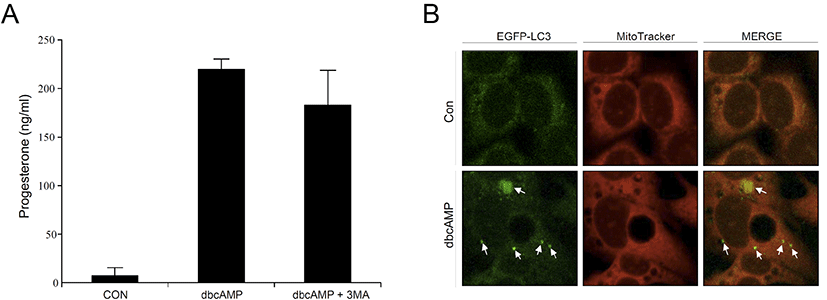
To confirm our findings in primary rat Leydig cells, we also used MA-10 cells, the mouse Leydig cell line, in this experiment. When dbcAMP was treated with MA-10 cells, progesterone (P4) levels were significantly increased in the cell culture medium. However, P4 levels tended to decrease in the presence of 3-MA, but the difference was not statistically significant (Fig. 5A). This was consistent with the results of the in vitro rat Leydig cell experiments. Finally, we transfected eGFP-LC3 into MA-10 cells to obtain representative morphological evidence of increased autophagy. As shown in Fig. 5B, dbcAMP-treated cells showed an increased amount of intense and punctate green fluorescence compared to the control, indicating that increased autophagic activity is required for steroidogenesis to a certain extent in Leydig cells.
Ultrastructural studies have demonstrated that autophagy is active and occurs spontaneously in Leydig cells (Tang et al., 1988; Yi & Tang, 1995). Autophagic deficiency in aged Leydig cells has been suggested to be associated with decreased steroidogenic activity (Li et al., 2011). The possible involvement of autophagy in T cell synthesis in Leydig cells has been demonstrated using autophagy-deficient Leydig cells (Gao et al., 2018). This study suggests that autophagy promotes cholesterol uptake into Leydig cells by eliminating the Na+/H+ exchanger regulatory factor 2, thereby corroborating with previous findings. However, these factors cannot explain the occurrence and basic cellular function(s) of autophagy, particularly in immature Leydig cells. Furthermore, based on our results, the autophagy pan-inhibitor 3-MA did not suppress hCG- or dbcAMP-stimulated steroid hormone (T or P4) production in primary adult Leydig cells or MA-10 cells.
Taken together, we believe that although autophagy participates in steroidogenesis and enhances steroidogenic efficacy of Leydig cells, it may not be a decisive cellular process for steroidogenesis, specifically in the mature Leydig cells. Recently, selective autophagy, a category of autophagy involved in the degradation of specific targets (e.g., damaged organelles, aggregated proteins, or invading bacteria), has been implicated in cellular quality control processes (Lamark & Johansen, 2021; Vargas et al., 2023). Therefore, it is important to elucidate the precise and specific roles of selective autophagy in Leydig cell steroidogenesis in the future.