
INTRODUCTION
For stem cell researchers, establishment of new human embryonic stem cell (hESC) lines and development of culture conditions for maintenance of the established cell lines efficiently and stably are fundamental areas of research, requiring further development and detailed investigations. In addition, recent studies have examined the clinical applications of stem cells from cell therapy products in regenerative medicine, highlighting the importance of securing hESCs with clinical grade quality (Hovatta, 2006; Tannenbaum et al., 2012). Thus, several groups have attempted to establish hESC lines, remove animal-derived components from culture conditions, and maintain xeno-free conditions. Reports on the establishment of new hESC lines and successful culture under xeno-free culture conditions have been published (Ellerström et al., 2006; Crook et al., 2007; Ilic et al., 2012; Tannenbaum et al., 2012), and many efforts have been made to develop appropriate stem cell culture protocols for the use of hESCs in clinical applications, from establishment and culture to freezing and thawing Skottman et al., 2007).
Since the first establishment of hESC in 1998 (Thomson et al., 1998), hESCs have been cultured in conditions using mouse derived fibroblasts, such as mouse embryonic fibro-blasts (MEFs) and STO as feeder cells. However, concerns remain regarding the potential risks of cell line contamination by unknown components secreted from these animal derived materials, therefore, studies have been performed to overcome these challenges (Soong et al., 2013). For example, studies have examined the replacement of mouse derived fibroblasts with human tissue-derived feeder cells, such as human foreskin fibroblasts, fetal muscle fibroblasts (Richards et al., 2002), umbilical cord derived mesenchymal stem cells (Ding et al., 2012), marrow stromal cells (Havasi et al., 2013), and placental cells (Park et al., 2011). Additionally, researchers have also attempted the development of extra-cellular matrix and xeno-free medium, which can be used in feeder-free conditions for cultures without feeder cells (Wang et al., 2013). There have also been continuous efforts to maintain hESCs efficiently and stably and establish new cell lines in the developed culture conditions.
Human amniotic fluid cells (hAFCs) have been used for prenatal genetic diagnosis to examine the possibility of fetal abnormalities, these cells can be easily obtained from pregnant women through second midtrimester amniocentesis (Prusa & Hengstschlager, 2002). Although few studies have charac-terized the various types of cells found in amniotic fluids, cells in amniotic fluids have been shown to be capable of differentiating into three germ layer cells. Thus, these cells have the potential for applications as materials for studies of neuronal differentiation or as stem cells (Prusa & Hengstschlager, 2002; Kook et al., 2006). Amniotic fluid derived stem cells have intermediate characteristics between hESCs and adult stem cells and express markers such as CD29, CD90, and CD105, which are expressed in bone marrow derived mesenchymal stem cells. These cells also express markers of the undifferentiated state of hES cells, including Oct-4, Nanog, SSEA-4, and TRA1-81 (Bajek et al., 2014). In particular, Oct-4, a representative marker for undifferentiated hESCs, has been shown to be expressed in 90% of amniotic fluid-derived stem cells (Prusa et al., 2003). Moreover, hAFCs have been shown to have higher reprogramming efficiency for establishment of induced pluripotent stem cells (iPSCs) than somatic cells (Galende et al., 2010). In addition, in studies using hAFCs as feeder cells, Kim et al. (2004) successfully cultured hESCs using hAFCs as feeder cells instead of mouse derived fibroblasts, which are more commonly used. Other reports have also described the maintenance of the undifferentiated state of hESCs using human amniotic fluid cell-derived stem cells as feeder cells without the addition of basic fibroblast growth factor (bFGF) (Ma et al., 2014), and new hESC lines have been established using amniotic fluid-derived mesenchymal stem cells as feeder cells (Soong et al., 2013).
In the current study, we examined whether clinical grade hESCs could be cultured in xeno-free medium without animal derived materials and under culture conditions using humanized extracellular matrix and hAFCs as feeder cells.
MATERIALS AND METHODS
The hESC line used in the present study was SNUhES32, a clinical grade cell line established at the Institute of Reproductive Medicine and Population of the Medical Research Center at Seoul National University. H1 from WiCell Research Institute were used as a control. Human amniotic fluid cells were used as feeder cells to maintain the undifferentiated state of hESCs.
hAFCs were stored frozen in vials in a liquid nitrogen (LN2) tank and thawed in T75 flasks coated with CELLStart (Invitrogen). Cells were cultured in medium containing 90% Dulbecco’s modified Eagle medium (DMEM; Invitrogen) and 10% fetal bovine serum (FBS; Hyclone) using an incubator (37°C, 5% CO2) for 3 days. To inactivate hAFCs, cell growth was suppressed by treatment with mitomycin C (Sigma) for 2.5 h, and the cells were then transferred to a CELLStart-coated 35-mm culture dish at a density of 2.0×104 cells/cm2.
SNUhES32 is a clinical grade cell line established in xeno-free medium using human foreskin fibroblasts as feeder cells, and H1 were used as control. For the culture of SNUhES32 and H1 using hAFCs as feeder cells, DMEM/ F12 (Invitrogen) was used as a basic medium and was supplemented with 15% KO-SR XenoFree (Invitrogen), 8 ng/mL bFGF (Invitrogen), 0.25 mM P/S (Invitrogen), 0.1 mM NEAA (Invitrogen), and 0.1 mM β-mercaptoethanol (Invitrogen). The cells were subcultured by the mechanical transfer method using a Pasteur pipette once a week for maintenance.
For Neu5GC test, H1 was maintained on mitomycin C-treated STO (CRL-1503; ATCC) feeders in DMEM/F12 with 20% KO-SR, 4 ng/mL bFGF, 0.25 mM P/S, 0.1 mM NEAA, and 0.1 mM β -mercaptoethanol (Sigma).
In order to examine whether the hESCs cultured using amniotic fluid cells as feeder cells maintained their own properties, immunocytochemistry (ICC), reverse transcription polymerase chain reaction (RT-PCR), and flow cytometry were used to analyze expression/activity of alkaline phosphatase (AP), a cell surface marker for the undifferentiated state of hESCs, and the expression of pluripotency markers, i.e., Oct-4, SSEA-4, TRA1-60 and TRA1-81.
After 5–6 days of subculture, SNUhES32 and H1 were washed with dPBS (Invitrogen) and fixed with 4% paraformaldehyde (PFA; Usb). AP expression was examined by staining using an Alkaline Phosphatase diagnostic kit (Sigma) according to the manufacture’s instructions.
Medium was removed from hESCs after 5–6 days of subculture. Cells were then washed with dPBS, fixed with 4% PFA, washed with dPBS containing 0.05% Tween-20 (Sigma) and 3% bovine serum albumin (BSA; Sigma) at a 1:1 ratio, and incubated at 4°C for at least for 12 h. Primary antibodies for Oct-4 (Santa cruz), SSEA-4 (Millipore), TRA1-60 (Millipore), and TRA1-81 (Millipore) were diluted 100-fold in 1% BSA and 0.1% Triton X-100 (Sigma) solution and added to the fixed cells, followed by incubation at 4°C for at least for 12 h. The cells were then washed with dPBS, mixed with secondary antibodies, Alexa Fluor 488-labelled donkey anti-mouse IgG (Molecular Probes), Alexa Fluor 594-labelled donkey anti-mouse IgG (Molecular Probes), or Alexa Fluor 594-labelled donkey anti-rabbit IgG (diluted 1:100; Molecular Probes) in a solution containing 1% BSA and 0.1% Triton X-100, as was used for primary antibodies. Cells were then incubated under dark conditions at room temperature for 1 h. Finally, nuclei were stained with DAPI (Molecular Probes), and the cells were observed under a fluorescent microscope.
RNA was extracted from hESCs using an RNeasy Mini Kit (Qiagen) according to the manufacture’s instructions. Reverse transcribed to cDNA using M-MLV Reverse Transcriptase (Invitrogen), and subjected to PCR to determine the expression of markers, including Oct-4, Nanog, Rex1, and Sox2. The sequences of primers used in this study are shown in Table 1.
Cell colonies were isolated from hESCs using a Pasteur pipette, transferred to microtubes, and washed with dPBS. hESCs were disassociated into single cells by treatment with accutase (Invitrogen) and centrifuged at 1,000 rpm for 3 min. After addition of 4% PFA and fixation at room temperature for 30 min, cells were centrifuged at 1,000 rpm for 3 min to isolate cells and mixed with primary antibodies targeting Oct-4 and SSEA-4 (diluted 1:100 in a solution containing 1% BSA and 0.1% Triton X-100). Cells were then incubated for 3 h and centrifuged at 1,000 rpm for 3 min. Similar to primary antibody treatment, the cells were treated with secondary antibodies, Alexa Fluor 488-labelled donkey anti-mouse IgG, or Alexa Fluor 594-labelled donkey anti-rabbit IgG (diluted 1:100), incubated under dark conditions at room temperature for 1 h, and centrifuged. An optimal volume of dPBS was then added to the cell pellet to resuspend, and the cells were subjected to analysis using a flow cytometer (FACSAria; BD Bioscience).
After hESCs culture using hAFCs as feeder cells without animal derived components, a flow cytometer was used to determine whether Neu5Gc (Biolegend), an animal derived component, could be detected. Samples were prepared as described for the flow cytometry analysis of markers for undifferentiated cells after incubation with primary anti-bodies targeting Neu5Gc, followed by incubation with Cy5- conjugated anti-chicken (Millipore) secondary antibodies. Cells were then analyzed using a flow cytometer (FACSCalibur; BD Science).
RESULTS
SNUhES32 and H1 were cultured for up to 10 passages while maintaining the undifferentiated state using hAFCs as feeder cells. During these 10 passages, the cultured hESCs maintained their own properties. For hAFCs used as feeder cells, only the fibroblast types shown in Fig. 1(A) were cultured and used. Both SNUhES32, which were established from human foreskin fibroblasts, and H1, which were established from MEFs, adapted well to hAFCs without complications, and round single layers, typical of hESC colonies, were observed (Fig. 1B and C).
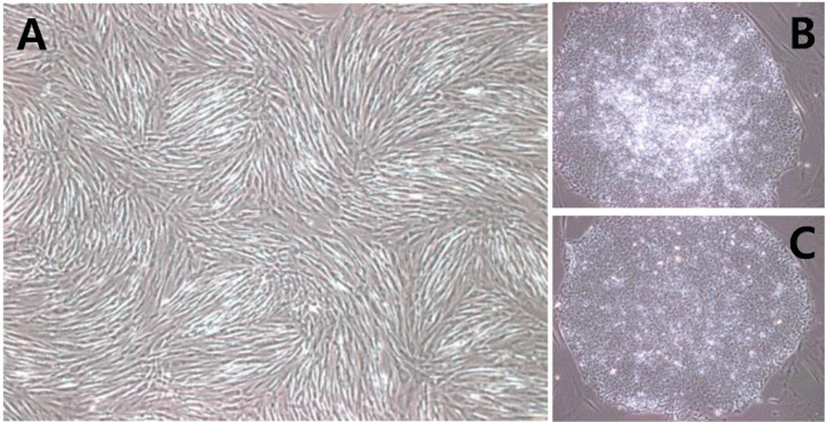
In this study, we attempted to replace materials that were generally used for hESC cultures with humanized materials in order to minimize exposure of hESCs to animal derived components during culture. Thus, hAFCs were used as feeder cells instead of mouse derived fibroblast culture. Culture dishes were coated with CELLStart, a humanized extracellular matrix, instead of 0.1% gelatin, and hESC medium was replaced with a medium containing KO-SR XenoFree. Under the xeno-free culture environment, both SNUhES32 and H1 were able to adapt to the new culture conditions.
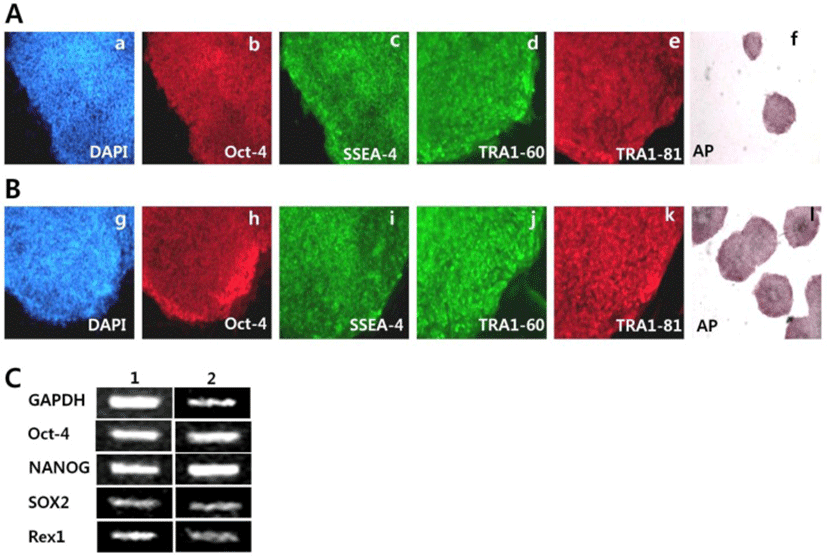
For characterization of SNUhES32 and H1 cultured on hAFCs, we tested that SNUhES32 and H1 at 8 passages, had AP activity, as determined using an AP staining kit. These data indicated that the undifferentiated state was maintained (Fig. 2f, i). In addition, immunocytochemistry (ICC), RT-PCR, and flow cytometry were used to test the expression levels of pluripotency markers for undifferentiated cells. The expression levels of cell surface markers, including SSEA-4, TRA1-60, and TRA1-81, and the transcription factor Oct-4 were determined using ICC staining and observation under a fluorescent microscope. Positive staining was detected in all cases (Fig. 2a–e, g–k). In addition, RT-PCR also demonstrated the expression of pluripotency markers of hESCs, including Oct-4, Nanog, Sox2, and Rex1, as shown in Fig. 2C.
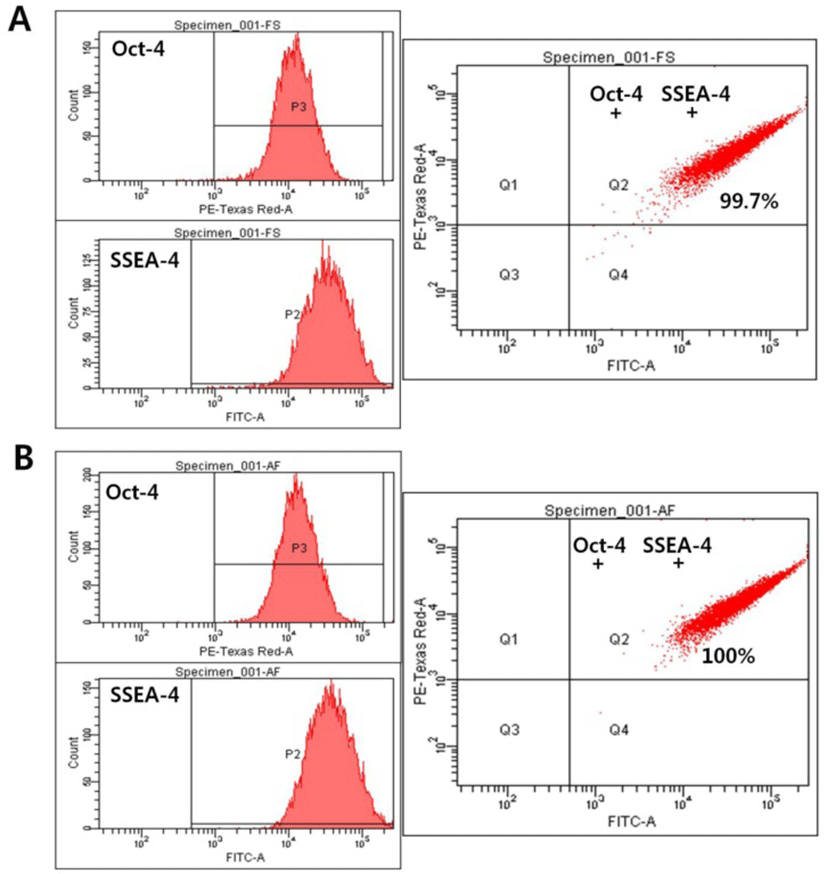
To further examine the expression of markers of the undifferentiated state, the expression levels of Oct-4 and SSEA-4 were quantitatively analyzed using flow cytometry. SNUhES32 cultured either in hAFCs or in human foreskin fibroblasts were analyzed for detection of markers, including Oct-4 and SSEA-4, by fluorescent staining combined with flow cytometry. No differences in the expression levels of Oct-4 or SSEA-4 were detected between the two experimental groups, with positive responses of up to 100% (Fig. 3).
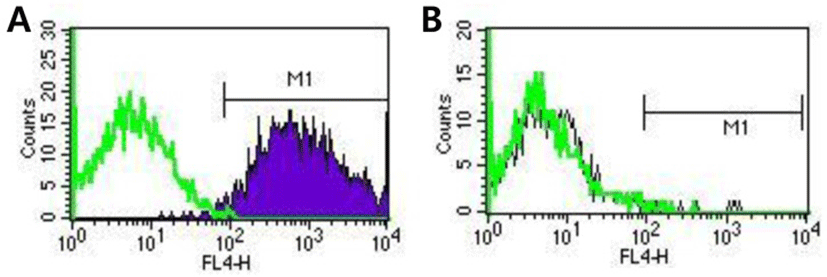
Finally, we investigated contamination of cultures with Neu5Gc by flow cytometry analysis using specific antibodies in order to examine whether animal derived materials were present after using the above described cell culture conditions. Our results showed that SNUhES32 grown in hAFCs were not contaminated with Neu5Gc (Fig. 4B). In contrast, Neu5Gc was detected in H1 cultured in STO (Fig. 4A).
DISCUSSION
Under currently used research protocols, medium containing mouse derived fibroblasts and FBS is generally used for culture of hESCs for research purposes. While these culture conditions are efficient for maintaining the undifferentiated state of hESCs, they are not suitable for use in clinical applications owing to risks of infection by various viruses and pathogens from the feeder cells and media used and the risks of contamination with other unknown components (Hovattta, 2006; Mallon et al., 2006). Therefore, researchers have attempted to reduce the use of animal derived materials and have focused on the development of new xeno-free culture conditions to completely exclude these materials. For example, human pluripotent stem cells (hPSCs) have been successfully cultured using human derived feeder cells (Richards et al., 2002; Kim et al., 2004; Park et al., 2011; Ding et al., 2012; Havasi et al., 2013; Soong et al., 2013; Ma et al., 2014), and clinical grade hESC lines have been established and cultured in xeno-free environments (Ilic et al., 2012; Tannenbaum et al., 2012). From these studies, some popular products have been commercialized for widespread use (Villa-Diaz et al., 2013).
In the present study, we aimed to determine whether SNUhES32 and H1, two hESC lines, could grow and maintain the undifferentiated state under new culture conditions removal animal derived materials. hAFCs were used as feeder cells, replacing mouse derived fibroblasts. These hAFCs were successful as feeder layers for both cell lines. SNUhES32 and H1 adapted well to medium containing KO-SR XenoFree rather than KO-SR and to xeno-free culture conditions using culture dishes coated with humanized CELLStart rather than pig-derived gelatin. SNUhES32, used as the experimental group, showed no distinct morphological changes compared with SNUhES32 cultured with human foreskin fibroblasts as a control, and characterization of pluripotency markers also showed that these cells maintained the properties of hESCs. Although we only examined the expression of markers of the undifferentiated state in this study, and we still used FBS during hAFC culture, we expect that hESCs will be able to be cultured in improved culture conditions in which FBS is replaced with human serum.
Based on currently published literature, mouse derived fibroblasts, materials secreted from fibroblasts, and animal derived materials, such as FBS, have immunological risks and risks of infection by animal viruses or prions, it is difficult to qualitatively assess such risks due to the presence of other unidentified materials (Ilic et al., 2009). For example, some xenotransplantation materials, such as Neu5Gc, were detected when hESCs were co-cultured with animal derived fibroblasts and in medium containing animal derived serum components. These materials are known to cause problems, including immune rejection and infection, during later transplantation (Martin et al., 2005). Hence, in the current study, we examined whether hESCs were contaminated with Neu5Gc, which is commonly detected under conditions in which animal derived feeder cells or medium containing animal-derived materials are used. Flow cytometry analysis with anti-Neu5Gc antibodies showed that the SNUhES32, grown on hAFCs, was free from Neu5Gc infection.
However, even in human derived feeder cells, used as a replacement for animal derived materials, the risks in clinical applications need to be broadly tested, including basic screening for diseases and viral infection in the donors (Stacey et al., 2006). Several factors should be tested for in human derived materials or cells to be used as biological medicines (Cobo et al., 2005), including human immunodeficiency virus (HIV), hepatitis (A, B, C, and E), human cytomegalovirus (hCMV), T cell leukemia virus (hTLV), herpes viruses (herpes simplex virus, Epstein-Barr virus, HV-6, HHV-7, and HHV-8), human polyoma virus, and parvovirus (B19 and TTV). In addition, it is essential to check for infections based on newly discovered, high risk infectious diseases, including Creutzfeldt-Jakob disease, a nonviral infectious disease, and severe acute respiratory syndrome (SARS).
For studies on clinical grade hESCs, the Reubinoff team reported the establishment of a clinical grade hESC line in xeno-free, GMP grade human derived feeder cells in 2012. This research team established and cultured clinical grade hESCs in xeno-free culture conditions using human derived feeder cells, including umbilical cord, foreskin, and fetal muscle cells that were cultured in a GMP grade clean room. They also verified the safety of all materials used in culture through quality management at each experimental step. In addition, they monitored steps required for verification of hESCs to be used as cell therapy products in the future, including tissues used as feeder cells, screening of embryo donors, establishment of hESCs, culture, freezing, characterization, and stem cell banking, and they established guidelines for ethical and systemic limitations and regulation of experimental methods in detail for clinical applications, providing a solid foundation for clinical grade studies (Tannenbaum et al., 2012). Based on these stem cell investigations, internationally standardized guidelines and regulations have been developed for individuals countries, including safety tests for quality assurance of basic cell cultures of hESCs and cell lines, and for institutional conditions according to various guidelines at the clinical stages required for hESCs to be approved as medical products.
Recently, studies in hPSCs, including hESCs, have focused on the next stage in clinical application, necessitating compatibility of cell therapy products with clinical application, the final goal of stem cell studies. Therefore, in addition to these changes, efficient culture conditions must be established for growth of hESCs by reducing animal derived components and replacing them with humanized or human derived materials in the basic cell culture environment. Studies should also be conducted with xeno-free and feeder-free environments that culture without feeder cells. In addition to improvement of culture conditions, quality management of cultured hESCs and standard procedures, including detection of microorganisms, viruses, and animal derived materials, for identification of inappropriate harmful substances need to be developed and standardized for production of clinical grade hESCs. This is the next preparatory step for the application of hESCs as cell based therapy in regenerative medicine.