
INTRODUCTION
Primary cell cultures are vital tools for the functional evaluation of biological substances and characterization of specific biological activities (Wang et al., 2020). Despite vast differences in the physiologies of mammals and fish, primary cell culture protocols for fish are often based on mammalian culture conditions with only minor modifications (Ager-Wick et al., 2018).
Primary culture of fish cells has been used as an ideal model for various studies, such as substrate utilization (Hasoon, 2018), xenobiotic metabolism, thermal stress, and monitoring of aquatic pollution (Wang et al., 1985; Ghosh et al., 1995). Recently, techniques have been developed for primary culture of fish cells, but the optimal protocols and conditions necessary for disaggregation, seeding, maintenance, and culture of cells have varied (Hasoon, 2018).
Moreover, using several organ or tissue from olive flounder, several cell lines have been developed: flounder gill (FG)-9307 cells (Tong et al., 1998), hirame natural embryonic cells (HINAE) (Kasai & oshimizu, 2001), flounder spleen and gill cells (Kang et al., 2003), flounder embryonic cells (Chen et al., 2004), and flounder brain cells (Zheng et al., 2015). Establishing various useful cell lines from olive flounder is important for basic research and biotechnological applications.
Primary cell culture is a sufficient method frequently used to study the cellular properties and mechanisms of isolated cells in a controlled environment (Ager-Wick et al., 2018). However, due to insufficient information on universally applicable cell culture conditions and protocols, well-described cell culture methods for marine fish are still limited.
Here, we investigated the conditions suitable for long-term maintenance of a primary embryonic cell culture. The cell line was also characterized chromosomal abnormalities, growth rate, and transfection capability in the present study.
MATERIALs AND METHODS
Embryonic cells were obtained by primary cell culture following methods in our previous study (Kim et al., 2018). Briefly, blastula-stage embryos from olive flounder were collected after 8 h in seawater at 18°C post-fertilization and prepared as follows. For embryonic cell harvest, 60–70 embryos were treated with antibiotics (1×), washed with DPBS (Gibco, Gaithersburg, MD, USA), and homogenized. The chorion membranes and cell debris were eliminated using a 40-μm cell strainer. For the single cells collection, the homogenate was centrifuged at 229×g for 15 min at 20°C, and supernatant was discarded. After several washes with growth medium (GM), the cells were moved to GM in a fresh cell culture flask (surface area: 25 cm2) (Corning, Corning, NY, USA).
Leibovitz’s L-15 complete GM (L-15; Gibco), supplemented with fetal bovine serum (FBS; Gibco), antibiotic–antimycotic (AA; Gibco), and fish serum (FS) from olive flounder, and growth factor (GF) were added to conduct the subculture according to the methods reported in our previous study (Kim et al., 2018). To prepare the FS, blood samples were collected from the caudal vein of three olive flounder and allowed to clot at 4°C for 4 h. After centrifugation, the serum was moved into new flash tubes, heat-inactivated in a water bath at 56°C for 30 min, filter-sterilized (0.2 μm), and stored at −20°C until use. FS concentration was determined via Warburg–Christian assay using a NanoVue spectrophotometer (GE Healthcare, Chicago, IL, USA) (data not shown).
Olive Flounder Embryonic Cell 18, named OFEC18-FGBC (FGBC8), was cultured at 20°C in an incubator with an air atmosphere, and the medium was changed every 2–3 days. To conduct the subculture, culture medium was prepared as GM (L-15 medium containing 10% FBS and 2% AA) with 1% FS and 0.1% GF (basal fibroblast GF [bFGF]; Corning). Upon reaching 90% confluence, the cells were subcultured at a ratio of 2:3 following our previous study (Kim et al., 2018) with minor modification. Briefly, FGBC8 cells were washed twice with GM (without FS and GF) and dissociated in trypsin-ethylenediaminetetraacetic acid (trypsin-EDTA [TE]; Gibco) for 3 min at room temperature and centrifuged at 330×g for 5 min at 20°C. The TE solution was removed, and single cells were suspended with fresh culture medium and transferred to a 25T cell culture flask (Corning).
For cryopreservation of embryonic cells, cultured cells were harvested with Bambanker stock solution (Lymphotec, Fuyuki, Japan).
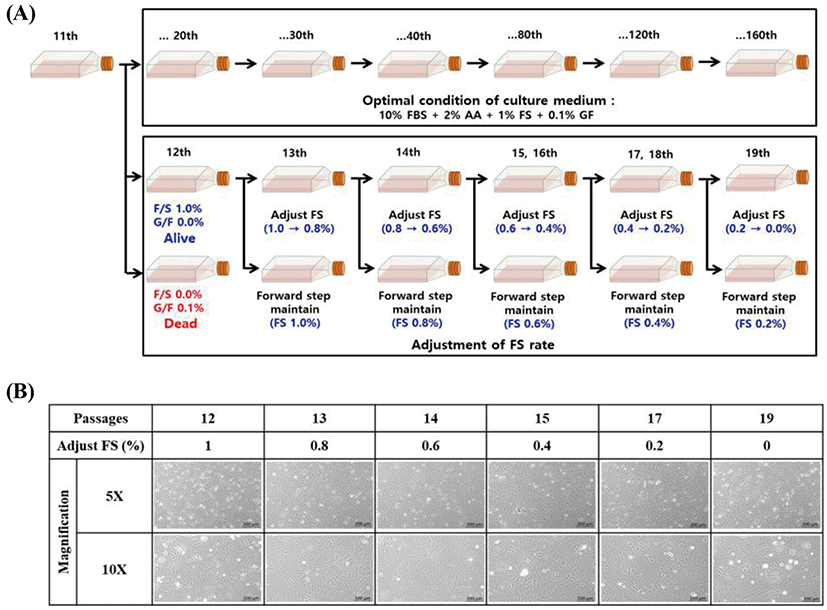
The effect of adjusting medium supplements was investigated to explore the optimal conditions for long-term maintenance of the embryonic cells. Cell culture medium was used as described above. When exchanging the cell culture medium in the subculture, FS was gradually decreased from 1.0% to 0%, while GF was decreased from 0.1% to 0%. The survival, growth, and morphology of FGBC8 cells were then visualized under a fluorescence microscope (Axio Vert A1; ZEISS, Oberkochen, Germany).
To measure cell growth, we used cell proliferation assays and general cell counting methods. First, a proliferation assay was conducted using a Premix WST-1 Cell Proliferation Assay System (Takara, Kusata, Japan) following a previously established method with minor modifications (Kim et al., 2013). The FGBC8 cells were adjusted to 6×104 cells/well in a 24-well plate (Corning) and incubated at 20°C for 4 days. Blank controls with GM alone were also included.
For the proliferation assay, 10 μL of WST-1 reagent was added to each well following incubation. The absorbance was determined using a Victor 3 microplate reader (PerkinElmer, Waltham, MA, USA) at a test wavelength of 450 nm and a reference wavelength of 690 nm.
For general cell counting, the FGBC8 cells were suspended in trypsin-EDTA for 4 min and centrifuged for 5 min at 330×g at 20°C. Trypsin-EDTA was then replaced with GM (1 mL). Cells were counted daily using a Bright-Line hemocytometer (Sigma-Aldrich, St. Louis, MO, USA).
Doubling time was calculated using the linear part of the growth curve as follows: doubling time = duration × log (2) ÷ [log (final conc.) – log (initial conc.)]. At the same time, the morphology of FGBC8 cells was observed using an IncuCyte S3 Live-cell Analysis System (Sartorius, Bohemia, NY, USA) for 4 days.
Data were analysed by one-way analysis of variance (ANOVA) using SPSS version 19 software (SPSS, Chicago, IL, USA). All samples were analysed in triplicate and the results are reported as the mean±SD.
FGBC8 cells were used for chromosome analysis following a previously published method (Kim et al., 2018) with slight modifications. Briefly, cells were treated with 1 μg/mL colchicines (Sigma-Aldrich) for 2 h 30 min at 20°C, harvested by scraping the flask using a sterile cell scraper (length: 290 mm, blade: 20 mm), suspended in 0.075 M KCl, and incubated for 20 min at room temperature. The KCl was discarded, and 4 mL methanol:acetic acid (3:1) fixative solution was added gently but rapidly using a glass Pasteur pipette (230 mm; Volac, Orwell, UK). The cells were incubated at room temperature for 30 min and 2× fixative solution was added. The cells were dropped onto a fixative solution-treated slide glass and stained for 8 min with 8% Giemsa (Gibco). Lastly, chromosomes of metaphase cells were visualized using a microscope (DE/EM6000B; Leica, Wetzlar, Germany) with oil-immersion optics (Merck KGaA, Darmstadt, Germany) at 1,000× magnification.
To determine the transfection efficiency of the FGBC8 cells, the transfection assay was conducted according to the methods reported in our previous study (Kim et al., 2018). FGBC8 cells were seeded at a density of 7×104 per well in 24-well plates at 20°C. Cell monolayers (80% confluent) were transfected with pEGFP-c1 plasmid DNA and an expression plasmid using a JetPEI kit (Polyplus, New York, NY, USA) according to the manufacturer’s instructions. Each well contained 500 ng pEGFP-c1 plasmid DNA in 150 mM NaCl (final volume: 50 μL) and 2 μL JetPEI reagent in 150 mM NaCl (final volume: 50 μL). Green fluorescence signals were detected under a fluorescence microscope (Axio Vert A1; ZEISS) at 48 h post-transfection.
RESULTS
A new olive flounder cell line called OFEC18-FGBC (FGBC8) was developed by primary cell culture of blastula-stage embryos following methods in our previous study (Kim et al., 2018). To explore the conditions required for long-term maintenance, we produced other embryonic cell lines under various conditions using the same culture methods; however, all newly developed embryonic cell lines died (data not shown) except the FGBC8 cells. The first subculture was also achieved. Morphologically, the FGBC8 cells were composed primarily of epithelial-like cells (Fig. 1). To date, FGBC8 cells have been subcultured for >160 passages over ~830 days.

We investigated the effect of adjusting FS to elucidate the ideal conditions for long-term maintenance. Although FS was gradually decreased from 1.0% to 0%, there was no effect on the survival of the tested cells (Fig. 2A). Conversely, when GF was decreased from 0.1% to 0.0%, the cells died. However, when cells adjusted to 0% FS were continuously subcultured (Fig. 2B), the survival rate gradually decreased until all were dead at 29 passages (data not shown). Therefore, we concluded that FS is a crucial supplement for the long-term survival and maintenance of comparable morphology of primary embryonic cells.
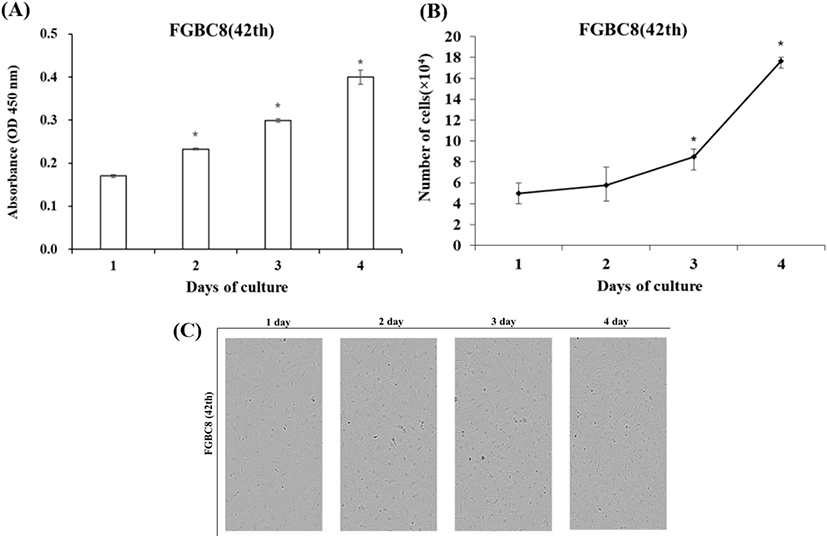
FGBC8 cells grew evenly attached to the well surfaces within the 24 h after seeding. Cell growth was assessed by cell proliferation assays and counting cell numbers with a hemocytometer. During the growth period, the number of cells increased, and the cells grew to confluence. Cell growth performance is shown in Fig. 3A and B. The FGBC8 cell density was 5×104 mL on day 1 and peaked at 17.7×104 mL on day 4, with a doubling time of 73.8 h. The cell morphology is shown in Fig. 3C.
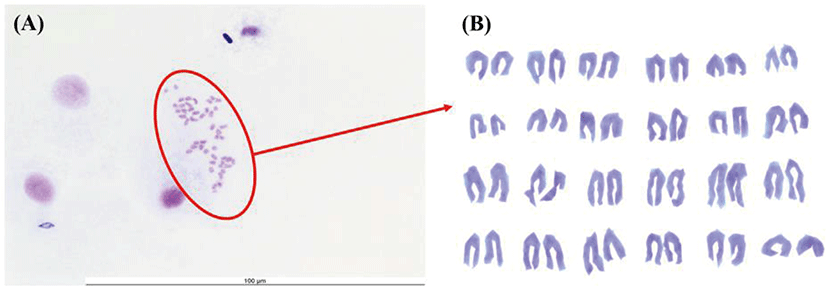
Chromosome morphology of FGBC8 is shown in Fig. 4. The chromosome numbers of the FGBC8 cells were assessed at passage 49. The number of chromosomes in metaphase FGBC8 cells was 48.
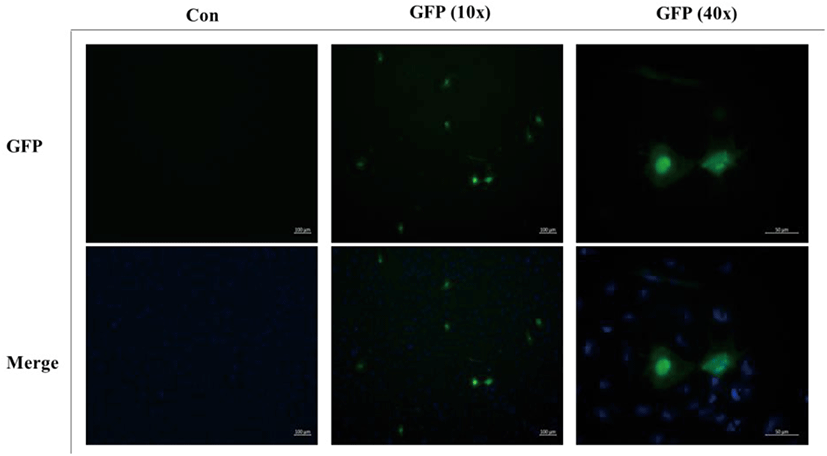
To determine the transfection efficiency and gene expression of the FGBC8 cells, cells were transfected with plasmid pEGFP-c1 using JetPEI reagent. FGBC8 cells transfected with pEGFP-c1 exhibited a strong green fluorescent signal at 48 h after transfection. The transfection efficiency of FGBC8 cells was approximately 10%–15% (Fig. 5). Thus, FGBC8 cells are useful for exogenous gene expression, which is important for both basic research and biotechnological applications.
DISCUSSION
In vitro cell culture is a vital tool in molecular biological research, providing an excellent model system for addressing research areas ranging from normal cellular physiology to drug screening and carcinogenesis (Ager-Wick et al., 2018).
Primary cells, isolated directly from animal tissue using enzymatic and/or mechanical methods, are often considered more biologically relevant than cell lines because the biological response may be closer to that in vivo (Ager-Wick et al., 2018). Protocols for preparing primary cell cultures should be optimized for each species and cell type of interest to mimic the characteristics to which a cell is adapted and obtain physiologically meaningful results (Ager-Wick et al., 2018).
In this study, novel olive flounder embryonic cell lines in vitro culture was developed and characterized in terms of their proliferation and transfection ability. Additionally, to establish the ideal conditions for long-term maintenance of primary embryonic cells, the effect of adjusting FS was evaluated.
In general, the media routinely used in fish cell culture are Glasgow MEM, Hank’s MEM (HMEM), and L-15 (Lakra et al., 2011). Amino acid-rich nutrient media, such as L-15, that do not require CO2 buffering have been successfully used with fish cell lines; thus, CO2 incubators are not necessary, and cells can be grown conveniently in any undisturbed area. Due to this advantage, more than 80% of cell lines established after 1994 have used L-15 medium (Wang et al., 1995). In this study, L-15 medium and FS were useful for primary cell culture and promoted in vitro growth of olive flounder embryonic cells, as reported in other fish species, e.g., Japanese sea bass, red seabream, and turbot Scophthalmus maximus (Chen et al., 2003a; Chen et al., 2003b; Chen et al., 2004). Here, we demonstrated that FS in media is essential for primary cell culture of flounder embryonic cells. Our results can be used as a reference guide for primary embryonic cell cultures of other fish species.
The development of embryonic cell lines is a difficult process using primary cell culture methods. The methodology outlined in this study demonstrates that embryonic cells isolated from blastula-stage flounder embryos can survive and retain comparable morphology in subculture for a long time. Furthermore, FGBC8 primary embryonic cells will allow further analyses of expression of immune-related genes using mitogens such as lipopolysaccharide, Poly I:C, flagellin, and interferon-γ and will further our basic understanding of immune responses. Further study of FGBC8 characteristics will allow full exploitation of the potential of this new embryonic cell line, and we encourage use of our method to explore and develop it as a model.