
INTRODUCTION
How maintain or improve the fertility of male or female is one of the main interestings in development and reproductive biology. The methods for preservation of sperm, oocyte, embryo and reproductive tissue has been developed recently. Oocyte cryopreservation has more wide clinical implications than embryos freezing procedure (Wennerholm, 2000) because women who have no partner or have the possibility to lose their ovarian function due to surgery, chemotherapy or radiotherapy can store their oocytes for future use. Cryopreservation of the excess oocytes can avert the repeated ovarian stimulation and oocyte collection in women undergoing in-vitro fertilization (IVF) or can be a source for oocyte donation. It also gives more chance for women to achieve their social goal and provide more reproductive choices because oocyte storage allows women to preserve the fertility for the future. Oocyte cryopreservation therefore has the potential to be an important adjunct to ART in human and other mammals. However, there has been the relatively limited preservative of mature oocyte (Veeck, 2003; Borini et al., 2004) compared with that of embryos (Mandelbaum et al., 1998).
Mature oocytes stay in metaphase block during secondary meiotic division in mammals including human. At this stage, the chromatins are condensed and become chromosome, and made specific connection with the metaphase II (MII) spindles. A spindle apparatus is a dynamic conglomerate of microtubules and associated structural proteins, acting to coordinate cytokinetic and karyokinetic events essential for normal chromosome segregation. First meiotic division is completed by cooperation of cytoskeletons, especially microfilaments and microtubules. Cytoskeleton is a key factor to get the successfully competent oocytes. Until sperm entry, the matured oocytes stay in metaphase block. This block is accomplished by stable spindle and other regulators for cell cycle. As oocytes transit into metaphase, microtubules change from radial arrays to an organized barrel-shaped bipolar structure containing a blend of dense material at either pole known as microtubules organizing centers (MTOCs).
The most important things are keeping the structural stability of both spindle and chromosomes for developmental competence. Unfortunately, many reports, however, have demonstrated that cryopreservation of oocytes may cause depolymerization and disorganization of spindle microtubules (Pickering & Johnson, 1987; Vincent et al., 1989; Aman & Parks, 1994). Oocytes are exposed to many stresses including thermal, mechanical and chemical stress during process of cryopreservation and caused the lost of survivability (Meryman, 1971; Mazur et al., 1972). In fact, the process of cryopreservation of meiotic spindles is leading to impairment of fertilization of such oocytes and the growth of embryos (Aman & Parks, 1994; Eroglu et al., 1998).
There are two methods that are used to cryopreserve the mamalian oocytes, slow-freezing and vitrification (Bernard & Fuller, 1996). High survival rates of human oocytes following slow-freezing have been achieved with sodium depleted medium and/or elevated sucrose concentrations (Fabbri et al., 2001; Bianchi et al., 2005; Borini et al., 2006; Stachecki et al., 2006). However, despite the increase in survival rates, implantation rates are still low; between 12-14% per transferred embryo (Boldt et al., 2006; Oktay et al., 2006). Recently, vitrification has been widely used for cryopreservation of ovine, equine, murine, rabbit, bovine, and porcine embryos at all stage of embryonic development, including blastocyst stages, and oocyte. It is suggested that vitrification may be more effective than slow-freezing (Kuleshova & Lopata, 2002), resulting in improved oocyte survival and pregnancy rates (Yoon et al., 2003; Katayama et al., 2003; Lucena et al., 2006).
Vitrification is a process that produces a glasslike solidification of living cells that completely avoids ice crystal formation during cooling and warming (Al-Hasani et al., 1986; Diedrich et al., 1988). However, a major concern of vitrification relates to the use of higher concentration of membrane-permeable cryoprotectants (CPA) in the suspending solution, such as dimethyl sulfoxide (DMSO), 1,2-propanediol (PROH), and/or ethylence glycol (EG). It has been known that exposure time to the high concentration of cryoprotectants during vitrification is one of the main factors influencing the oocytes. Therefore in this study, the stability of meiotic organelles was examimed to evaluate the vitrification influences on the survival of mature oocyte. In addition, cryoinjury of oocytes was detected at DNA level during vitrification.
MATERIALS AND METHOD
All animals involved in this study were approved by the Animal Care Committee and studies were conducted for the Care and Use of Laboratory. To get MII stage oocyte, the female mice were injected with 5 IU of pregnant mares serum gonadotropin (PMSG; Folligon, Intervet) followed by injection with 5 IU of human chrionic gonadotropin (hCG) in 48 hrs later (i.p.). After 15 h of hCG injection, the mice were sacrificed and the oviducts were dissected and placed into a dish containing Quinn's Advantage™ Medium with HEPES (Sage Biopharma, San Clemente, CA; cat. no. 1023) supplemented with 1.0 mg/mL bovine serum albumin (BSA) (Sigma, A8022). The cumulus-oocyte complexes (COC) were released by tearing the ampullae of the oviduct. The cumulus mass were removed using both hyaluronidase (80 IU/mL) (Sigma, H3506) and mechanical force using a fine glass pipette. The denuded oocytes were washed several times with Quinn's Advantage™ Medium with HEPES supplemented with 1.0 mg/mL BSA at RT and undergone for next examination.
Oocytes were cryopreserved by vitrification methods as described by Kuwayama and his colleagues (2005) with minor modifications. The solution for vitrification and warming was PBS containing cryoprotectants and 10% serum protein substitute. The equilibration solution consisted of 7.5% ethylene glycol (EG; Sigma E9129) and 7.5% dimethyl sulfoxide (DMSO; Sigma D5879). The vitrification solution consisted of 15% EG and 15% DMSO, 0.5 M sucrose (S; Sigma S7903). The oocytes were placed in 7.5% ethylene glycol EG and 7.5% DMSO for 5, 10 and 15 min. Then those were transferred into the vitrification solution (15% EG + 15% DMSO + 0.5 M sucrose). After 40~60 sec of exposure in the vitrification solution, oocytes were loaded onto straw (Fig. 1) and directly plunged into liquid nitrogen (LN2).

After being stored in LN2 for at least 1 week, warming was accomplished holding the frozen straw for 10 sec at 37°C. Oocytes were sequentially rehydrated in PBS containing 1 M sucrose for 1 min at 37°C and 0.5 M sucrose for 3 min at RT and rinsed twice in PBS for 5 min, respectively. And then the oocytes were transfered back into the culture medium and cultured for an additional 1 hr.
Oocytes were defined as having morphologically survived if the oocytes possessed an intact zona pelludia and plasma membrane and refractive cytoplasm. They were counted and recorded.
Tubulin and chromatin were stained by immunofluorescent staining, as described previously (Jack et al., 2007). Briefly, after warming the oocytes were incubated for 1 hr at 37°C in 5.0% CO2 incubator. Then the warmed oocytes were fixed in 4% paraformaldehyde for 5 minutes. After fixation, the oocytes were washed extensively in PBS and blocked overnight at 4°C in blocking medium (PBS, supplemented with 0.02% NaN3, 0.2% non-fat dry milk, 2% goat serum, 2% BSA and 0.1 moL/L glycine). After rinsing in PBS, the oocytes were incubated with β-tubulin monoclonal antibody (Sigma F2043) diluted 1:150 in PBS at 4°C overnight. After then, oocyte washed several times with PBS and incubated with fluorescein isothiocyanate (FITC)- conjugated anti-mouse IgG (Sigma F5387) diluted 1:150 in PBS for 60 min at 37°C. After washing with PBS for several times, the samples were mounted onto a slide under a cover slip in the Vectashield mounting medium (Vector Laboratories, Burlingame, CA), containing 4,6-diamidino-2-phenylindole (DAPI).
Morphologically intact oocytes underwent comet assay to detect cryoinjury at DNA level. The individual oocytes were employed for comet assay that was carried out as previously described (Singh et al., 1988). All products for comet assay were obtained from Trevigen, Inc (Gaithersburg, MD). Quantitative analysis of DNA damage included the counting the number of oocytes with the presence of a comet tail.
RESULTS
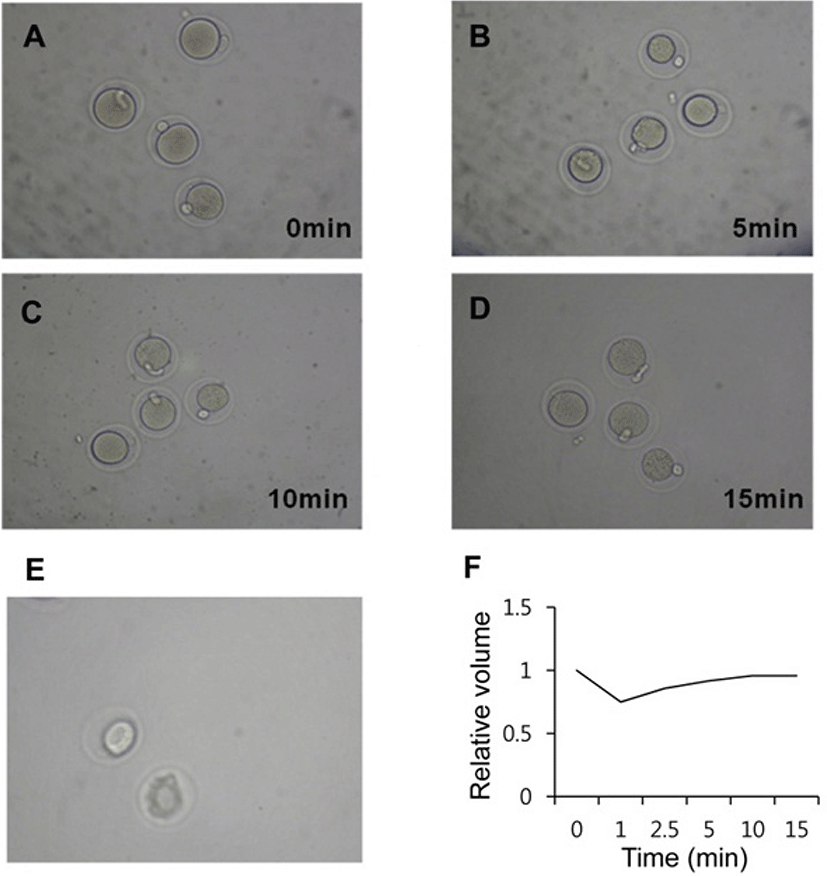
The volumatric changes were recored during vitrifycation (Fig. 2). At the time of equilibration, within 1 min after expose to dehydration solution the oocytes were shrunk (Fig. 2B) and got equilibration from 10 min (Fig. 2C, D). After treatment with vitrification solution for 40 sec, there was dramatic shrinking of the equlibrated oocytes (Fig. 2E).
A total 445 oocytes were vitrified. After warming and washing, oocytes were assessed for cryosurvival according to the criteria mentioned in Materials and Methods (Fig. 3). Survival rates of the recovered oocytes were higher than 90% and there was no difference between experimental groups. MII oocytes after vitrification and warming were morphologically indistinguishable compared to them before cryopreservation. The recovered oocytes had normal-appearing zona pellucidae, intact polar bodies, normal perivitelline spaces, intergral plasma membranes, and normal-appearing evenly granular cytoplasm. After 1 hr of culture, the survival rates were 97.1% (102/105) and 93.5% (174/186) and 95.5% (147/154) for 5, 10, and 15 min, respectively (Table 1). From those results, the device for vitrification and time point were good to keep the viability of oocytes.
| Time (min) | No. of vitrified oocytes | Survival rate (%) |
|---|---|---|
| 5 | 105 | 102/105 (97.1 %) |
| 10 | 186 | 174/186 (93.5 %) |
| 15 | 154 | 147/154 (95.5 %) |
Morphological analysis is not enough to prove the stability of competency of matured oocytes. So we analyzed the stability of spindle and chromosome. The spindle morphology and chromosomal patterns of vitrified oocytes of 1 hr incubation in 5% CO2, in air at 37°C. The majority of the recovered oocytes maintained the normal meiotic spindle morphology and chromosome alignment (Fig. 4).
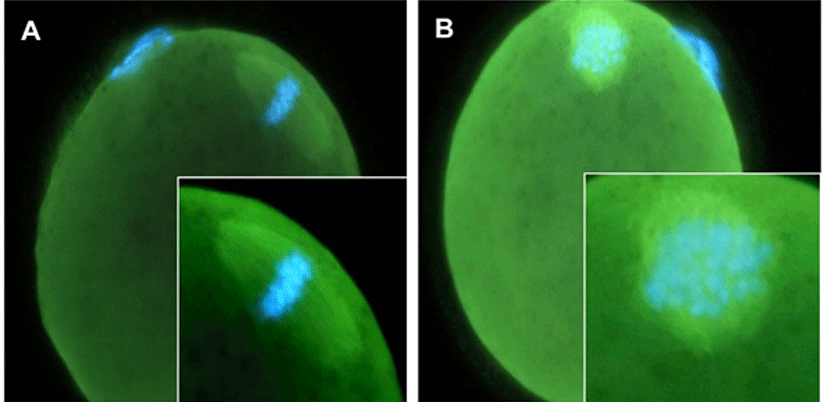
The rates of oocyte having normal spindle and chromosomal configuration were 74.3%, 81.1% and 66.3% in 5 min group, 10 min group, and 15 min group, respectively. In control group which were collected after superovulation, the rate of normal spindle and chromosomal arrangement was 87%. Compared to the control group, there were no significant difference in 5 min and 10 min group, but there was a significant decrease in 15 min group (Table 2).
| Equilibration time | No. of oocyte | Spindle and chromosome configuration (%) |
|
|---|---|---|---|
| Normal | Abnormal | ||
| Control | 69 | 60 (87.0) | 9 (13.0) |
| 5 min | 77 | 57 (74.3) a | 20 (25.7) |
| 10 min | 90 | 73 (81.1) | 17 (18.9) |
| 15 min | 83 | 55 (66.3) a,b | 28 (33.7) |
The 10 min equilibration gave more stability signifycantly than the other groups. Compared with control and 10 min group, the stability did not change (Table 2). However, the stability of spindle and chromosomal configuration was significantly decreased in 5 min and 15 min group compared to 10 min group.
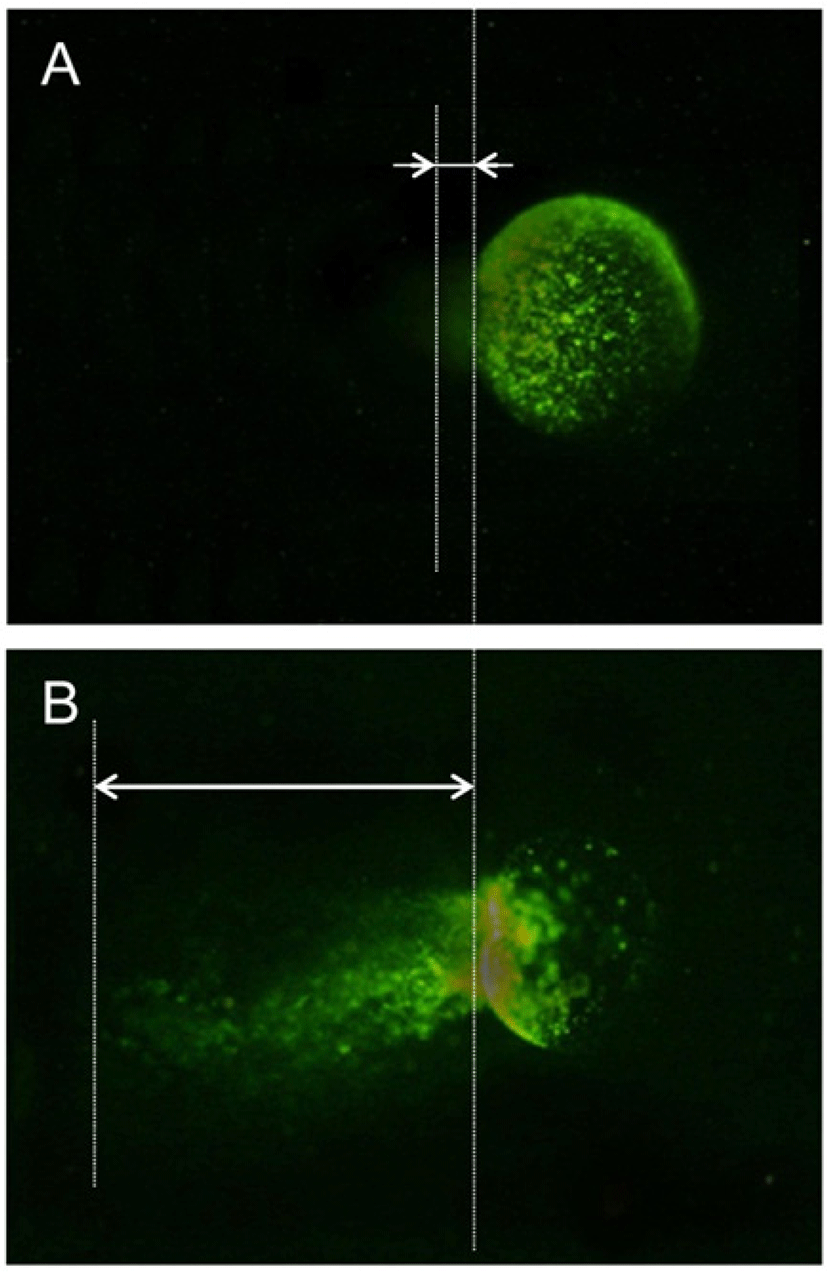
One of the well established method for the analysis of the chromosomal stability is comet method and it was applied after morphological analysis (Fig. 5). In control group, the oocytes which had comet tail were 15.3% but the percent of comet tail was increased dramatically in the cryopreserved oocytes (Table 3).
| Equilibration time | No. of oocytes | No. of oocytes with comet tail (%) |
|---|---|---|
| Control | 13 | 2 (15.3) |
| 5 min | 14 | 6 (42.9) a |
| 10 min | 16 | 7 (43.8) a |
| 15 min | 15 | 12 (73.3) a, b |
As shown in Table 3, the percentage of oocytes with comet tails was significantly higher (P<0.05) in 15 min group compared to 5 min exposure group. However, there were no difference in the percentage of oocytes with comet tails between 5 min exposure group oocytes and 10 min exposure group.
DISCUSSION
A major advantage of vitrification is the elimination of mechanical damage caused by intra- or extra-cellular ice crystals and potential reduction of chilling damage by shortening the exposure to suboptimal temperatures. However, vitrification is a result of high cooling rates associated with high concentrations of cryoprotectants. If used incorrectly, these cryoprotectants can be toxic. Therefore, vitrification necessitated the reduced and precise exposure times and rapid movement of cells through solution.
An important problem with oocyte cryopreservation is the variable survival rate (Chen, 1986; Porcu et al., 1997; Boldt et al., 2003; Fosas et al., 2003). Modified slowfreezing methods have improved survival rates (Fabbri et al., 2001; Fosas et al., 2003; Bianchi et al., 2005; Borini et al., 2006; Stachecki et al., 2006), but as yet it is still unclear as to which method provides the best overall success. However, it is suggested that vitrification has advantages in varios points compared with slow-freezing. In this study, the survival rates of recovered oocytes are almost same between groups and are higher than 93%. The suvival rate was higher previously reports which employed vitrification and other freezing methods (Huang et al., 2008).
At the time of ovulation, the oocyte is arrested at MII stage of meiosis with the chromosomes aligned at the equatorial plate of the meiotic spindle and the first polar body extruded. The completion of meiosis depends on the presence of intact spindle microtubules to achieve normal segregation of the chromatids. Spindle microtubules consist of polymerized tubulin, the major component of spindle microtubules, in equilibrium with the free tubulin pool within the oolemma (Vincent & Johnson, 1992). The dynamic equilibrium between polymerized and free tubulins in mammalian oocytes is extremely sensitive to temperature change (Zenses et al., 2001). Cooling of mouse oocytes causes tubulin to undergo depolymerization and results in the disappearance of microtubule organizing centers (Magistrini & Szollosi, 1980; Webb et al., 1986; Pickering & Johnson, 1987). In addition, exposure of human and animal oocytes to cryoprotectant can induce spindle alterations and chromosomal anomality (Van Elast et al., 1988; Sathananthan et al., 1988; Pickering, 1990). Based on the morphological analysis of spindle microtuvules and chromosome, normal rate of spindle and chromosome configuration was significantly high in 10 min equlibration time group compared with the other groups.
Surprisingly the chromosomal breakdown was severe in the recovered oocytes in all experimental groups. The chromosomal stability was kept well in 10 min equilibration group compared with the other groups. The 10 min equilibration group showed best condition compared with the other groups, but the rate was less than 56%. Therefore, to improve the stability of chromosome, more fine analysis about the effects of physical stress on oocyte during vitrification is needed to define the optimal condition, it is suggested that the optimal equilibration time to get competent oocyte in mouse is 10 min.
The survival rate was higher than those of the previous reports independently to the equilibration times. In addition, the normal rate of spindle and chromosome configuration was improved by changing the equlibration time. However, the stability of DNA was not dramatially improved. Based on these results, it is suggested that the equilibration time is one of the key factors in successful ekeeping for competence of mature oocyte. Information acquired this study with mouse oocyte may provide insight into intracellular structural events occurring in human oocytes after vitrifycation and application for cryopresevation of human or other mammals oocytes.