
INTRODUCTION
Polycyclic aromatic hydrocarbons (PAHs) are carcinogenic compounds that arise from the incomplete combustion of organic substances and are abundantly present in tobacco smoke and tar (Hattemer-Frey & Travis, 1991; Hecht, 1999). PAHs, including benzo[a]pyrene (B[a]P), 3-methylcholanthrene, and 2,3,7,8,-tetrachlorodibenzo-p-dioxin, are specific inducers of drug-metabolizing enzymes such as CYP1A1, CYP1A2, and CYP1B1 in cells (Puga et al., 2002). PAHs can bind to the aryl hydrocarbon receptor (AhR), a member of the basic HLH (helix-loop-helix)-PER-ARNT-SIM (bHLH-PAS) family of transcriptional factors (Denison & Nagy, 2003; Ko et al., 2004). The ligand-AhR complex translocates to the nucleus, where it binds to the AhR nuclear translocator (Arnt) to specific cis-acting regulatory DNA promoter sequences known as AH-, dioxin-, or xenobiotic-responsive elements (AHRE, DRE, or XRE, respectively) (Ohtake et al., 2003).
Similar to other PAHs, B[a]P requires AhR for its action in cells (10). B[a]P can be converted to its metabolized forms [benzo[a]pyrene-7,8-diol (B[a]P-diol, DHD) and benzo[a]pyrene-7,8-diol-9,10-epoxide (BPDE)] by the activation of CYP1A1/CYP1B1 and epoxide hydrolase (EH) (Fig. 1). CYP1A1 metabolizes electrophilic metabolites that form DNA adducts and induces oxidative DNA damage, thereby causing mutations and initiation of carcinogenesis (Lewis & Parry, 2004). In addition, cytotoxicity-provoked cell death induced by B[a]P has been shown in several cell types, including hepa1c1c7 hepatoma cells (Ko et al., 2004), ovarian germ cells (Matikainen et al., 2002), bone marrow stromal-B cells (Allan et al., 2003), human macrophages (van Grevenynghe et al., 2004), Daudi human B cells (Salas & Burchiel, 1998), and RL95-2 human endometrial cancer cells (Kim et al., 2007).
During mammalian spermatogenesis, Sertoli cells in the seminiferous tubules of the testes play a pivotal role in supporting germ cell proliferation and differentiation (Siu & Cheng, 2004). Therefore, possible functional and/or structural disruptions of these cells after exposure to exogenous chemical substances can adversely affect the sperm generation process. In fact, it has been shown that mono-(2-ethylhexyl) phthalate and di-(2-ethylhexyl) phthalate, well-known plasticizers, negatively affect Sertoli cells and disrupt spermatogenesis (Raychoudhury & Kubinski, 2003). However, it remains uncertain whether B[a]P directly exhibits cytotoxicity in Sertoli cells or its metabolites show cytotoxicity. In the present study, we evaluated whether mouse testicular TM4 Sertoli cells are susceptible to the induction of cytotoxicity-mediated cell death after exposure to B[a]P in vitro. Furthermore, the potential requirement of AhR and CYP1A1 for B[a]P action was investigated in this cell type.
MATERIALS AND METHODS
Benzo[a]pyrene (B[a]P), Dulbecco’s modified Eagle’s medium nutrient mixture F-12 HAM, paraformaldehyde (PFA), propidium iodide (PI), Rhodamine 123, RNase A and anti-actin were purchased from Sigma-Aldrich (St. Louis, MO, USA). Benzo[a]pyrene-7,8-diol (DHD) and BPDE were from Midwest Research Institute in National Cancer Institute Repository (Kansas City, MO, USA). Fetal bovine serum, horse serum, penicillin-streptomycin and trypsin were purchased from Gibco (Thermo Fisher Scientific, Waltham, MA, USA). Antibodies for AhR and CYP1A1were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-cleaved caspase-3 and caspase-7 from Cell Signaling Technology (Danvers, MA, USA). Anti-actin from Sigma-Aldrich. The western enhanced chemiluminescence (ECL) detection reagent was purchased from Bio-Rad (Hercules, CA, USA).
TM4 cells (mouse Sertoli cell line; American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium nutrient mixture F-12 HAM containing 5% heat-inactivated fetal bovine serum, 2.5% heat-inactivated horse serum and 1.2 g/L sodium bicarbonate supplemented with 10 μg/mL penicillin-streptomycin. The cells were incubated at 37°C in a humidified incubator with an atmosphere of 5% CO2 and were exposed to B[a]P (0.1, 1, 10, 100 mM), DHD (1 μM) or BPDE (1 μM) when confluency reached 30%.
The cells were harvested, fixed with 95% ethanol for 24 h, incubated with 0.05 mg/mL PI and 1 μg/mL RNase A at 37°C for 30 min, and analyzed by flow cytometry, using the Epics XL system and analysis software (EXPO32 TM; Beckman Coulter, MI, USA). The cells belonging to the sub-G1 population were considered to be apoptotic cells.
The cells (5×105) were incubated with 5 μM Rhodamine 123 dye at room temperature for 30 min, washed and resuspended with PBS, and then the fluorescence [red (585/590 nm); green (510/527 nm)] was measured with a flow cytometer.
Whole-cell lysates were prepared by incubating cell pellets in lysis buffer [30 mM NaCl, 0.5% Triton X-100, 50 mM Tris-HCl (pH 7.4), 1 mM Na3VO4, 25 mM NaF, 10 mM Na4P2O7, protease inhibitor cocktail] for 60 min on ice. After the insoluble fractions were removed by centrifugation at 13,000×g at 4°C for 40 min, the supernatants were collected and protein concentration was determined with a BCA protein assay kit (Pierce Biotechnology, Woburn, MA). The same amounts of proteins (~50 μg) were subjected to SDS-PAGE and transferred onto a nitrocellulose membrane (Amersham Bioscience, Buckinghamshire, UK). The membranes were incubated overnight at 4°C with a primary antibody in Tris-buffered saline containing 0.05% Tween-20 [TBS-T (pH 7.4)] in the presence of 5% nonfat dry milk. After the membranes were washed in TBS-T, secondary antibody reactions were performed with an appropriate source of antibody conjugated with horseradish peroxidase. The signals were detected with ECL detection reagent in the LAS-4000 (Fuji, Tokyo, Japan). Actin was used as internal control for total cellular proteins.
Total cellular RNA was isolated from cultured cells by using a Trizol reagent (Invitrogen, Carlsbad, CA, USA). The complementary DNA (cDNA) was synthesized from 5 μg total RNA using oligo dT random primer (Promega, Madison, WI, USA) and MMLV RNase H-reverse transcriptase (Promega). Three μL of cDNA was subjected to PCR in a 30-μL reaction mixture [10X PCR buffer, 2.5 mM dNTP, Taq-polymerase 5 U (Promega), upstream and downstream primers]. The primers used were AhR (Abbott et al., 1999); forward 5’-CGC TGA AAC ATG AGC AAA TTG G-3’, reverse 5’-ACA GCT TAG GTG CTG AGT CAC AGG-3’. Thermal cycling conditions were 95°C for 1 min; 15 s at 54°C, 1 min at 60°C, 30 s at 72°C for 35 cycles, and 72°C for 30 min. The PCR products were analyzed by 2% agarose gel electrophoresis and visualized by ethidium bromide staining under UV illumination.
Harvested cells were attached to glass slides by cytospin centrifugation. The cells were fixed with 4% PFA, washed with PBS, and incubated with 0.2% Triton X-100. Cells were then incubated with the appropriate primary antibody in 1% bovine serum albumin at RT. For the secondary antibody reaction, cells were incubated with an appropriate fluorescence-conjugated secondary antibody at RT. Finally, the cells were mounted on glass slides and observed under a confocal microscope (LSM510, Carl Zeiss, Oberkochen, Germany).
Cellular proteins were isolated with lysis buffer consisting of 30 mM NaCl, 50 mM Tris-HCl (pH 7.6), 5% Triton X-100, and 100 mM PMSF. The enzymatic activity of CYP1A1 was measured by P450-Glo™ assay kits (Promega) as per manufacturer’s instruction manual. Briefly, isolated proteins (30 μg) were mixed with the 4X cytochrome P450/KPO4/substrate reaction mixture (CYP1A1; 0.5 pM CYP1A1 isozyme, 400 mM KPO4, 120 μM leuciferin-CEE), and 2X NADPH regeneration mixture (2.6 mM NADP+, 6.6 mM glucose-6-phosphate, 0.4 U/mL glucose-6-phosphate dehydrogenase, 6.6 mM MgCl2). The sample and the 4X cytochrome P450/KPO4/substrate reaction mixture were added to a 96-well plate. After preincubating the plate at 37°C for 10 min, the 2X NADPH regeneration mixture was added to each reaction. The plate was incubated at 37°C for 30 min, and the reconstituted leuciferin detection reagent was added. Again, the plate at RT was incubated for 20 min, and the luminescence recorded using a luminometer (Type392; Amersham Bioscience, Sweden).
RESULTS AND DISCUSSION
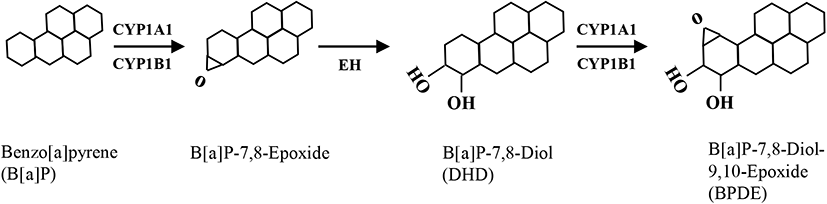
B[a]P can be classified as a potent carcinogen (Cole et al., 2003) as well as an endocrine-disrupting chemical, depending on the specific cell type (Brody & Rudel, 2003). In mammalian testes, Sertoli cells directly support spermatogenesis by secreting many bioactive substances such as growth factors, cytokines, and steroid hormones in response to follicle-stimulating hormone (Siu & Cheng, 2004). Therefore, if these cells are negatively affected by exposure to cytotoxic levels of xenotoxic chemicals, spermatogenesis can be seriously disrupted. In this study, TM4 Sertoli cells were exposed to various concentrations of B[a]P and its metabolites, i.e., DHD and BPDE. Flow cytometric analysis is commonly employed to evaluate cytotoxicity-mediated cell death because it is a quantitative and accurate method for measuring the rate of apoptosis in a cell (sub-G1) population (Chung et al., 2007a, 2007b; Kim et al., 2007). A dose-response (0.1–100 μM) treatment of B[a]P did not cause significant cell death at any concentration compared to that in the control (Fig. 2A). This cellular feature was not altered even with an extended duration of treatment (up to 72 h) at 10 μM B[a]P (Fig. 2B). Furthermore, TM4 cells did not undergo noticeable cell death following treatment with DHD, an intermediate metabolite of B[a]P generated by CYP1A1/1B1 and EH (Fig. 2C). However, BPDE, a final metabolite of B[a]P generated by CYP1A1 activation from DHD as a substrate, significantly induced cell death (Fig. 2D). These results indicate that TM4 Sertoli cells might be potentially deficient in AhR and/or CYP1A1 expression, which is required for the conversion of B[a]P to its genotoxic product BPDE through DHD. Consistent with these results, we have previously demonstrated that cellular defense mechanisms against B[a]P in testicular Leydig cells are associated with insufficient expression of AhR and CYP1A1 proteins (Chung et al., 2007a, 2007b). In contrast, cell types that are susceptible to apoptosis in response to B[a]P commonly retain abundant levels of AhR and CYP1A1/CYP1B1 proteins (Solhaug et al., 2004; Chung et al., 2007a, 2007b; Kim et al., 2007). Therefore, it is believed that sufficient expression of AhR and CYP1A1 is a prerequisite for the induction of cytotoxicity-mediated cell death by B[a]P.
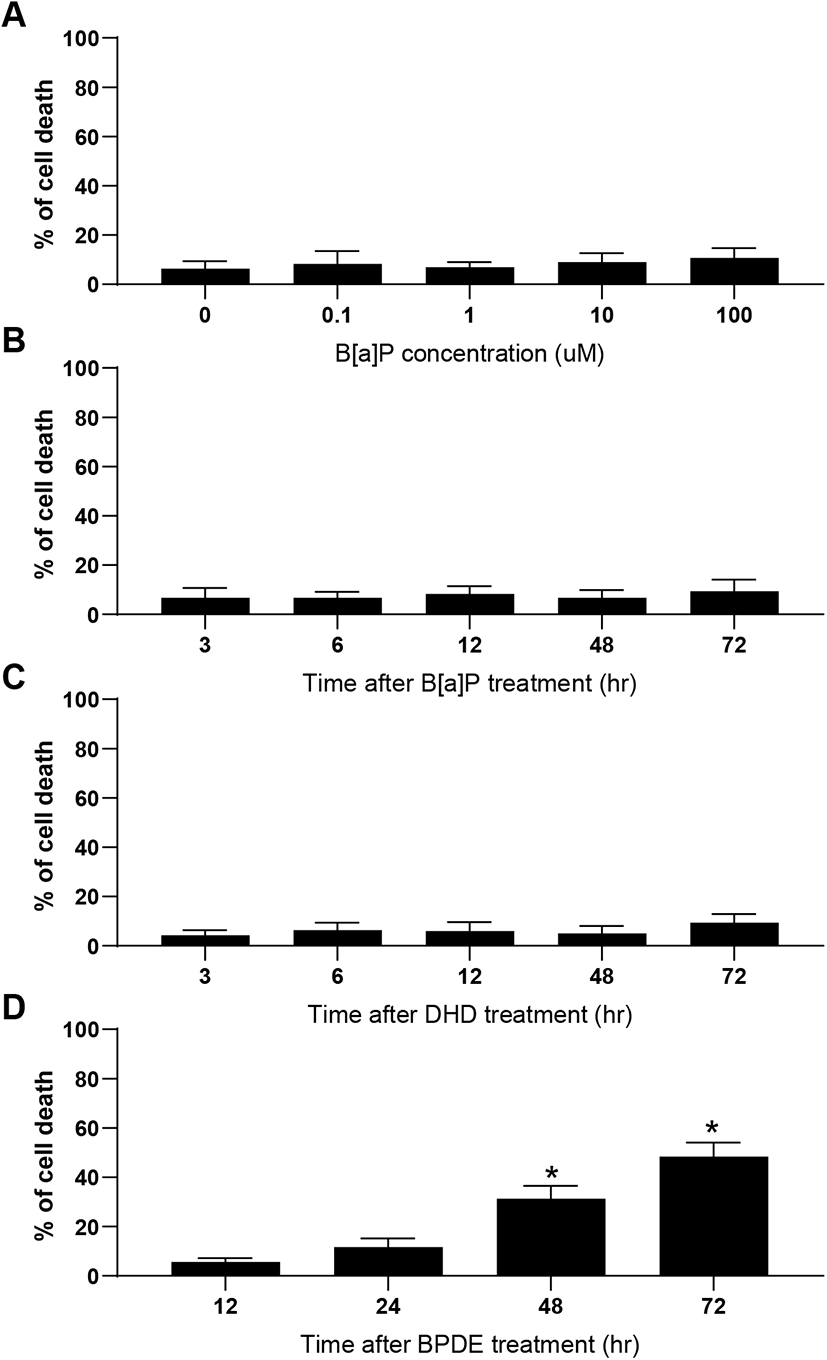
Cytotoxicity-mediated cell death determined by sub-G1 analysis of the cell cycle only represents the rate of apoptotic cell death induced by toxic agents. During apoptotic cell death, the activation of effector caspases (caspase-3 and -7) has been implicated in both death receptor-mediated and mitochondrial pathways (Shi, 2004). To confirm apoptotic cell death biochemically and biophysically, we analyzed the activation of caspase-3 and caspase-7 proteins and measured the mitochondrial membrane potential (MMP) in TM4 cells after exposure to B[a]P, B[a]P-7,8-diol, and BPDE. Consistent with the results obtained from the sub-G1 analysis, caspase-3 and caspase-7 activation [determined by the cleaved products of the effector caspases (caspase-3 and -7)] was remarkably observed in the BPDE-treated cells but not in the B[a]P- and B[a]P-7,8-diol-treated cells (Fig. 3A). These results were also confirmed at the cellular level using immunocytochemical observations (Fig. 3B). BPDE-induced apoptosis was accompanied by the depolarization of the mitochondrial membrane (Fig. 3C). In addition, cytochrome c release, mostly due to the decreased MMP, was monitored immunocytochemically in the BPDE-treated cells (Fig. 3D). These results indicate that if B[a]P can be converted to BPDE, BPDE-induced apoptosis in TM4 Sertoli cells involves the mitochondrial pathway.
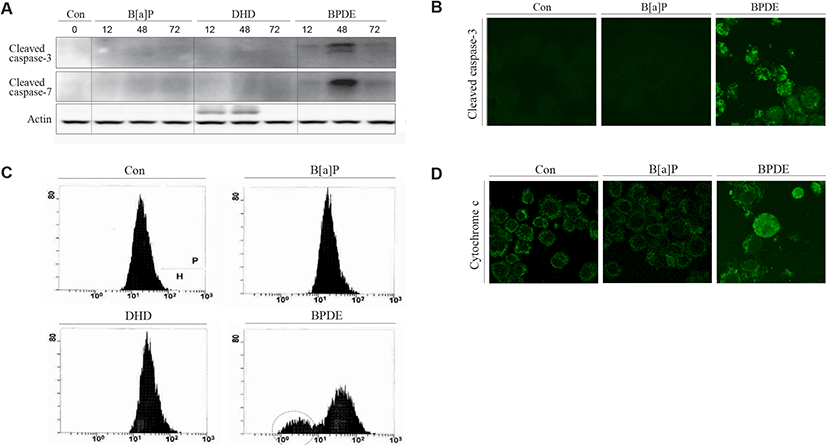
To determine why B[a]P is not able to exert its cytotoxic effect in TM4 cells, two decisive proteins (AhR and CYP1A1) associated with B[a]P signaling and metabolism have been investigated in TM4 cells. B[a]P can bind to AhR and translocate to the nucleus with Arnt, where the AhR-Arnt complex triggers the transcription of CYP1A1 via AhRE activation (Nebert et al., 2000). In the present study, western blot and RT-PCR analyses showed that AhR expression was almost undetectable in TM4 cells and that its expression was not altered after B[a]P treatment (Fig. 4A, 4B and 4C). This indicates that TM4 cells are nearly AhR-deficient. CYP1A1, a microsomal enzyme, plays a major role in the conversion of PAHs into genotoxic derivatives (Hattemer-Frey & Travis, 1991). In TM4 cells, the CYP1A1 protein and its activity were not detected (Fig. 5A, 5B and 5C). To confirm the absence of the CYP1A1 protein and activity in this cell type, we employed hepa1c1c7 cells as a positive control for CYP1A1 expression and activity after B[a]P treatment (Fig. 5A, 5B and 5C). From these results, it is clear that AhR may be a prerequisite for CYP1A1 expression in TM4 cells. Therefore, TM4 cells can be referred to as CYP1A1-deficient cells.
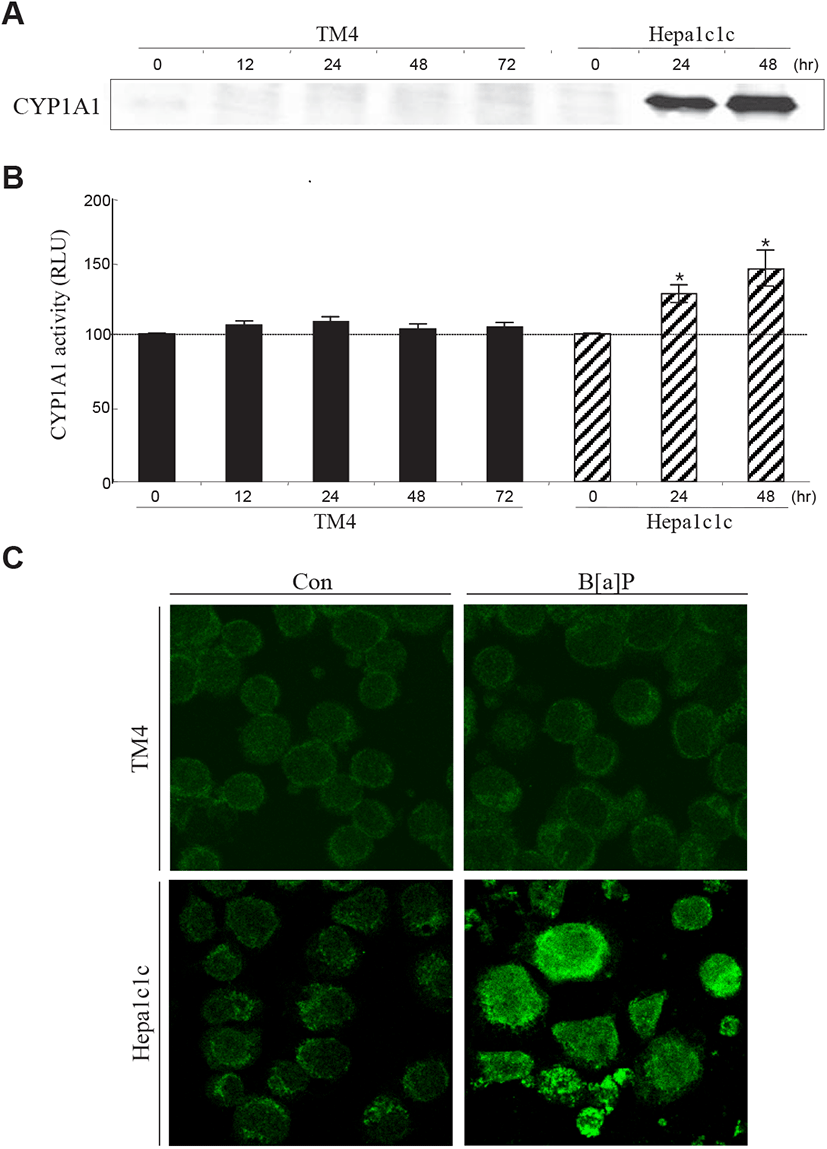
Thus, TM4 Sertoli cells are believed to have a rigid and protective cellular machinery against genotoxic agents. Although the characteristics of TM4 cells cannot be exactly identical to those of Sertoli cells in the testes, the fundamental responsiveness to exogenous toxic chemicals may be analogous. In conclusion, it is suggested that tolerance to B[a]P cytotoxicity is associated with insufficient AhR and CYP1A1 expression in testicular Sertoli cells.